
Structuring hybrid palladium nanoparticles in metallic monolithic reactors for continuous-flow three-phase alkyne hydrogenation†
Davide
Albani
a,
Gianvito
Vilé
a,
Mario Alejandro
Beltran Toro
b,
Rolf
Kaufmann
b,
Sharon
Mitchell
a and
Javier
Pérez-Ramírez
*a
aInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1, Zurich, CH-8093, Switzerland. E-mail: jpr@chem.ethz.ch
bEMPA, Uberlandstrasse 129, Dubendorf, CH-8600, Switzerland
First published on 15th July 2016
Abstract
Palladium nanoparticles modified with the hexadecyl-2-hydroxyethyl-dimethylammonium dihydrogen phosphate (HHDMA) ligand have been stabilised within the microchannels of a metallic monolith coated with a high-surface area γ-alumina layer. The stainless steel skeleton of the monolith was 3D printed by stereolithography. The washcoating protocol has been optimised in order to maximise the γ-alumina loading without blocking the microchannels. A battery of techniques has been applied to characterise the properties and three-dimensional organisation of the phases within the monolithic reactor from the macro- to the nanoscale including N2 sorption, X-ray diffraction, thermogravimetric analysis, nuclear magnetic resonance and infrared spectroscopies, X-ray tomography, and optical or electron microscopies. Evaluation of the catalyst performance in the flow hydrogenation of acetylenic compounds of different size and functionality demonstrates the high efficiency and stability of the structured catalyst. Particularly, the monolithic reactor retains the intrinsic selective character of the hybrid palladium nanoparticles, even at high temperatures and pressures (T > 343 K and P > 5 bar). This is attributed to the improved isothermicity of the catalyst bed, deriving from the high thermal conductivity of the metallic skeleton of the monolith. Overall, the work provides a general route to prepare monolithic reactors based on Pd-HHDMA nanoparticles, bridging the gap between hybrid nanomaterial and reactor engineering.
Introduction
Versatile synthetic strategies that synergistically couple facets of chemistry, process engineering, and materials science are steering innovations in diverse technologically-important fields, including heterogeneous catalysis.1 In the last decades, the emergence of hybrid nanoparticles, which combine inorganic and organic components at the molecular scale, has introduced an inexhaustible abundance of new catalysts with intriguing properties.2,3 For example, the development and commercialisation by BASF of a platform of supported palladium and platinum nanoparticles modified with the hexadecyl-2-hydroxyethyldimethyl ammonium dihydrogen phosphate (HHDMA) ligand (NanoSelect™) and supported on porous TiSi2O6 or activated carbon carriers has revitalised the field of hydrogenation catalysis, importantly applied for the manufacture of fine chemicals.4,5 This has demonstrated that the possibility to replace state-of-the-art Lindlar-type catalysts and react to major environmental and economic concerns depends on the ability to rationally design hybrid nanostructures with controlled surface composition.In the fine chemical industry, catalysts have traditionally been evaluated in batch mode. The main advantage of this type of reactor is its versatility when processing compounds in small volumes, since different operations can be carried out in a single vessel. However, it is now established that continuous flow in micro-reactors offers several advantages in terms of process intensification, safety, and sustainability. For this reason, our group has previously evaluated the performance of NanoSelect™ in random packed-bed micro-reactors.6–9 An interesting approach to further enhance the material functionality involves the organisation of the active phase into straight channel or otherwise macroporous (e.g., foam, gauze, or fibre-based) monolith-type structures.10 This can bring more controlled process conditions (pressure, residence time, temperature, etc.) and higher productivity in terms of activity and selectivity.12
Different strategies have been reported to anchor metal nanoparticles in monolithic reactors. Most commonly these involve the deposition of precursor salts followed by a high-temperature or solvent-assisted reduction treatment.11–17 The monolith may be pre-coated with a porous inorganic washcoat or functionalised with an organic layer18–27 to improve the adhesion of the active phase. An alternative approach would be to immobilise preformed ligand-modified metal nanoparticles onto suitably designed monoliths. Here, we demonstrate the effective stabilisation of Pd-HHDMA nanoparticles on the γ-Al2O3 coated surface of a steel monolith. The use of a metallic frame, in particular, can offer advantages to prevent the formation of hot spots, which are likely in highly-exothermic reactions occurring over a packed catalyst bed. The nano- and macroscopic properties of the resulting structured reactor are characterised by state-of-the-art techniques. The advantages of structuring Pd-HHDMA nanoparticles into monolithic reactors are demonstrated by comparing the performance of the structured reactors with respect to more conventional packed beds in the continuous-flow three-phase hydrogenation of various acetylenic compounds.
Experimental
Materials preparation
The monoliths were prepared by stereolithography using a Schaublin 28-CCN 3D printer. This method is well suited for the fabrication of miniaturised monoliths for microapplications and enables the accurate replication of complex geometrical features. A metal alloy (stainless steel 316L) was chosen for the monolith skeleton due to the enhanced thermal characteristics with respect to a ceramic body. The monoliths, featuring an external diameter (d) of 3.5 mm and a length (L) of 24.8 mm, contained 19 square channels with a side diameter (s) of 0.40 mm, and minimum wall thicknesses (w) of 0.25 mm. The metallic walls of the monolith were coated with a γ-Al2O3 layer, using a sol–gel method. The sol was prepared by mixing boehmite (Dispersal P2, Sasol), urea (Sigma-Aldrich, 99.9%), and 0.3 M nitric acid (Acros Organics, 60%), in a weight ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5, followed by vigorous stirring for 24 h. The urea served to moderate the viscosity of the sol while the nitric acid acted as a dispersant and surface modifying agent.11 The monolith was dipped into the sol for 60 s, withdrawn slowly from the solution, and dried by blowing compressed air (0.5 bar) through the microchannels. The washcoated monolith was finally dried at room temperature and calcined in static air at 723 K for 4 h, using a heating rate of 5 K min−1. Unless otherwise specified, this procedure was repeated three times. The synthesis of the Pd-HHDMA nanoparticles, which were kindly supplied by BASF, is reported elsewhere.8 Briefly, an aqueous solution of HHDMA (10 cm3, 30%, Sigma-Aldrich) was diluted in deionised water (300 cm3) and mixed with an aqueous solution of Na2PdCl4 (0.1 M, 5 cm3). The pH of the mixture was adjusted to 5 by addition of NaOH (12 M). The resulting solution was heated to 353 K and stirred at this temperature for 1 h, leading to the nucleation of colloidal metal nanoparticles of approximately 5 nm in diameter. The washcoated monolith was dip-coated in 1 cm3 of the resulting solution and dried at 333 K for 2 h (Fig. 1), yielding the sample coded Pd-HHDMA/Al2O3-m.
:1:5, followed by vigorous stirring for 24 h. The urea served to moderate the viscosity of the sol while the nitric acid acted as a dispersant and surface modifying agent.11 The monolith was dipped into the sol for 60 s, withdrawn slowly from the solution, and dried by blowing compressed air (0.5 bar) through the microchannels. The washcoated monolith was finally dried at room temperature and calcined in static air at 723 K for 4 h, using a heating rate of 5 K min−1. Unless otherwise specified, this procedure was repeated three times. The synthesis of the Pd-HHDMA nanoparticles, which were kindly supplied by BASF, is reported elsewhere.8 Briefly, an aqueous solution of HHDMA (10 cm3, 30%, Sigma-Aldrich) was diluted in deionised water (300 cm3) and mixed with an aqueous solution of Na2PdCl4 (0.1 M, 5 cm3). The pH of the mixture was adjusted to 5 by addition of NaOH (12 M). The resulting solution was heated to 353 K and stirred at this temperature for 1 h, leading to the nucleation of colloidal metal nanoparticles of approximately 5 nm in diameter. The washcoated monolith was dip-coated in 1 cm3 of the resulting solution and dried at 333 K for 2 h (Fig. 1), yielding the sample coded Pd-HHDMA/Al2O3-m.
 | ||
| Fig. 1 Key steps (1–5) in the stabilisation of hybrid Pd-HHDMA nanoparticles within the microchannels of a metallic monolith coated with γ-Al2O3. The length (L), diameter (d), channel side (s), and wall thickness (w) of the square-channel monolith are indicated. | ||
For reference purposes, the colloidal Pd-HHDMA nanoparticles were supported on a γ-Al2O3 carrier (sample coded Pd-HHDMA/Al2O3-p), which was derived from the washcoat applied to the monolith. In this case, the sol was dried at room temperature and calcined under equivalent conditions (i.e., 723 K for 4 h, using a heating rate of 5 K min−1). After synthesis, the Pd-HHDMA/Al2O3-p catalyst was dried overnight at 333 K. The commercial catalyst (coded NanoSelect, Strem Chemicals, ref: 46-1711), comprising Pd-HHDMA supported on a TiSi2O6 carrier, was used as received.
Materials characterisation
The palladium content in the catalysts was determined by inductively coupled plasma-optical emission spectrometry (ICP-OES) using a Horiba Ultra 2 instrument equipped with a photomultiplier tube detection. The monolith was dissolved in HNO3 (65%, Sigma-Aldrich), whereas the powdered catalyst was digested in aqua regia. The C, H, N, and P contents were determined by infrared spectroscopy using a LECO CHN-900 combustion furnace. Nitrogen isotherms were measured at 77 K in a Micrometrics TriStar II instrument, after evacuation of the samples at 393 K for 3 h. X-ray diffraction (XRD) patterns of the ground samples were measured on a PANalytical X'Pert PRO-MPD diffractometer, using Ni-filtered Cu Kα radiation (λ = 0.1541 nm). Data were recorded in the 2θ range of 5–70° with an angular step size of 0.05° and a counting time of 8 s per step. 27Al magic angle spinning nuclear magnetic resonance (MAS NMR) was performed on a Bruker Avance 400 spectrometer equipped with a 4 mm probe head and 4 mm ZrO2 rotor at 104.2 MHz and with a spinning speed of 10 kHz. Spectra were obtained using 1024 accumulation, 90° pulses with a pulse length of 2.4 μs, a recycle delay of 0.25 s, and with (NH4)Al(SO4)2·12H2O as reference. Temperature-programmed desorption of ammonia (NH3-TPD) was carried out in a Micromeritics Autochem II 2920 chemisorption analyser coupled with a MKS Cirrus 2 quadrupole mass spectrometer. The sample (0.1 g) was pretreated in He (30 cm3 min−1) at 673 K for 2 h. Afterward, 10 vol% of NH3 in He (30 cm3 min−1) was adsorbed three times at 473 K for 30 min, followed by purging in He (30 cm3 min−1) at the same temperature for 1 h. NH3 desorption was monitored in the range of 473–1073 K, using a heating rate of 10 K min−1. Infrared spectroscopy of adsorbed pyridine was performed in a Bruker IFS 66 spectrometer equipped with a liquid N2-cooled mercury–cadmium–tellurium detector. The self-supporting wafer (50 mg, 1 cm2) was evacuated at 10−7 bar at 673 K for 4 h. The sample was then saturated with pyridine vapour at room temperature, followed by evacuation for 1 h. The spectra were recorded in the 4000–650 cm−1 range, by co-addition of 32 scans at a resolution of 4 cm−1. Digital-reflected light microscopy images of the monolith exterior were obtained with a Leica DVM 5000 microscope equipped with a charge-coupled device camera and a tungsten halogen lamp. An X-ray computed tomography scan at micrometre resolution (micro-CT) of the whole monolith was undertaken on a custom-built X-ray micro-CT machine at Empa with a resolution of 5.4 μm. The high resolution was achieved via two practical strategies: (i) cutting the embedded monolith (LR White) into sections of approximately 0.06 mm thick, using a diamond wire saw; (ii) exploiting the point projection geometry of the imaging setup to obtain higher projection magnification. In addition, an image processing algorithm adapted from the seminal work of Paganin et al. was applied to each projection in order to increase to reconstruction quality.28,29 Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS) was conducted using an FEI Quanta 200F microscope operated at 5 or 20 kV, respectively. Scanning transmission electron microscopy (STEM) was undertaken using an FEI Talos S200 microscope operated at 200 kV and equipped with high-angle annular dark field (HAADF) and EDS detectors. The sections were further thinned to ca. 40 nm in thickness by dimple grinding and subsequently with a precision ion polishing system (Gatan). The ultrasonic adherence test was conducted by treating the monolith in toluene for 30 min, using an ultrasonic bath (Bandelin). The monolith was weighed before and after the test, after drying at 333 K for 3 h, and the weight loss was calculated with respect to the washcoat loading.30 Thermogravimetric analysis was performed in a Mettler Toledo TGA/DSC 1 Star system. Prior to measurement, the samples were dried in N2 (40 cm3 min−1) at 393 K for 1 h. The analysis was performed in air (40 cm3 min−1), heating the sample from 298 K to 1173 K, at a heating rate of 5 K min−1.Catalytic evaluation
The hydrogenation of 2-methyl-3-butyn-2-ol (Acros Organics, 99.9%), 1-hexyne (Acros Organics, 98%), 3-methyl-1-pentyl-3-ol (TCI, 98%), 3-hexyne (Acros Organics, 99%), and 4-octyne (Acros Organics, 99%) was carried out in a fully-automated flooded-bed micro-reactor (ThalesNano H-Cube Pro™), in which hydrogen (produced in situ via water electrolysis) and the liquid feed (introduced by an HPLC pump, Knauer) flow concurrently upward through the catalyst (catalyst particle size of 0.2–0.4 mm). In the case of the packed-bed reactor, apart from the catalyst, the cartridge was filled with an inert material in a weight ratio 1:1 (particle size = 0.2–0.4 mm); γ-Al2O3 (prepared in this manuscript), TiSi2O6 (Sigma-Aldrich, 99.8%), or SiC (Alfa Aesar, 99%). The residence time (τ) was calculated as the inverse of the liquid hourly space velocity (LHSV), as elaborated in the ESI.† Briefly, the liquid hourly space velocity in the packed bed was calculated as the ratio between volumetric flow rate of the liquid and the volume of the catalyst bed.31 For the monolith, the liquid hourly space velocity was calculated as the ratio between the volumetric flow rate of the liquid and the total volume of the channels.32,33 To prove that the reactions were conducted under kinetic control, we have estimated the influence of mass- and heat-transport phenomena on the reaction kinetics, using criteria based on measurable or observable quantities (e.g., Carberry number, Wheeler–Weisz criterion, extraparticle and intraparticle heat transfer criteria).34,35
The liquid feed contained 5 wt% of substrate dispersed in toluene (Fischer Chemicals, 99.95%) and 3 wt% of benzene (ABCR-Chemicals, 99%) as internal standard. To rule out the possible volatilisation of the internal standard during reaction, we have previously conducted blank tests using inert SiO2.6 A solution of the alkyne (5 wt%) and benzene (3 wt%) in toluene was prepared and fed through the cartridge, at different temperatures (303–363 K). The outlet feed from the reactor was collected and analysed by GC, monitoring the ratio between alkyne and benzene. This resulted in deviations lower than 1%. This finding is consistent considering the short residence time in the reactors (for the packed bed τ = 4.7 s and for the monolith τ = 4.2 s, details in the ESI†). The reactions were conducted at various conditions of temperature (303–363 K), pressure (1–8 bar), and liquid (1 cm3 min−1) and H2 (36 cm3 min−1) flow rates. The HPLC pump enabled the precise feeding of the substrate solution into the microreactor. The reaction products were collected employing a liquid handler (Gilson GX-271), after 10 min of steady-state operation, and analysed offline using a gas chromatograph (HP-6890) equipped with a HP-5 capillary column and a flame ionisation detector. The GC analyses were conducted using helium as a carrier gas (flow rate = 3 cm3 min−1); the column was kept at an initial temperature of 313 K for 0.5 min before ramping the temperature from 323 to 473 K with a heating rate of 10 K min−1. The conversion (X) of the reactant was determined as the amount of reacted alkyne divided by the amount of alkyne at the reactor inlet; the selectivity (S) to a given product was quantified as the amount of that compound divided by the total amount of reacted alkyne. The retention times of all products were assigned based on pure standards and the amounts formed were quantified using calibration curves with respect to the internal standard.
Results and discussion
Catalyst preparation
The steps applied to structure the Pd-HHDMA nanoparticles in the microchannels of a stainless steel monolith are illustrated in Fig. 1. Due to the nonporous nature of stainless steel, an alumina-based washcoat layer was applied to enhance the available surface area and improve the interaction with the hybrid nanoparticles. The washcoat was introduced in the monolith by dip-coating in a sol of AlO(OH), followed by drying and calcination. The loading and thickness of the washcoat layer was controlled by varying the number of dip-coating steps. Three repetitions were found to yield a reasonably uniform layer of ca. 50 μm thick (Fig. 2a and b), as shown by the stepwise decrease in the average free length (t) of the square microchannel from the starting value of 0.4 mm. Further dip-coating steps undesirably led to visible blockage of the microchannels. The non-linear variation in the washcoat thickness with the number of dip-coating steps reflects the poor controllability of applying washcoats based on a slurry-coating mechanism (nonporous support), due to the differences in adhesion that can be expected between the washcoat and the bare or pre-coated metal surface, which can lead to increased internal heterogeneity. An adhesion test was conducted to confirm the effective hardening of the γ-Al2O3 layer and strong interaction with the stainless steel wall. Only a minor weight loss (3 wt% with respect to the initial washcoat loading) was observed after the treatment, verifying the stability of the monolithic reactor. | ||
| Fig. 2 Average ‘free’ length (t) in the monolith microchannels estimated by image analysis of optical micrographs as a function of the number of dip-coating steps (a). The blue line represents the standard deviation. Optical micrographs of the monolith with square microchannels after different numbers of dip-coating steps (b). The inset shows the definition of ‘free’ length (t). | ||
Catalyst properties
Table 1 shows the bulk composition and textural properties of the alumina carrier derived from the washcoat and of the powder and monolith catalysts. To gain detailed insight into the properties of the washcoat layer, the structural transformations induced by the treatments applied during the catalyst preparation (i.e., the drying and thermal decomposition of the boehmite sol and the impregnation of Pd-HHDMA) were investigated over the powder support. As expected, analysis by XRD (Fig. 3a) confirms that upon high-temperature treatment the pseudoboehmite precursor, which initially exhibits all of the reflections of a well-crystallised boehmite (ICSD 93730), is completely transformed into a γ-Al2O3 (ICSD 66559) phase displaying broad reflections. 27Al MAS NMR studies were conducted to determine and assign different 4-, 5-, or 6-coordinate aluminium sites in the support (Fig. 3b). As expected, the pseudoboehmite precursor contains only 6-coordinate aluminium, evidenced by the single peak at 6.8 ppm, which is consistent with the presence of octahedral AlO6 species sharing edges and vertices within the structure.36 On the other hand, the γ-Al2O3 attained upon calcination displays an additional peak at 61 ppm indicative of the presence of 4-coordinate AlO4 sites. Moreover, the peaks are broadened and skewed (tailing to low frequencies) with respect to the precursor, implying a wider distribution of electric field gradients related to the increased disorder (e.g., due to the presence of defects) within the sample. | ||
| Fig. 3 (a) X-ray diffraction patterns and (b) 27Al magic angle spinning nuclear magnetic resonance spectra of the dried boehmite washcoat (green), the γ-Al2O3 carrier obtained upon calcination of the latter at 723 K (red), and of the Pd-HHDMA/Al2O3-p sample (blue). Vertical lines on the top and bottom axis in (a) show the reference patterns of the γ-Al2O3 (ICSD 66559) and boehmite (ICSD 93730), respectively. (c) NH3-temperature programmed desorption profile of the γ-Al2O3 carrier. The profile of TiSi2O6 is shown for reference (black). | ||
Characterisation by N2 sorption confirms the high (meso)porosity of the γ-Al2O3 powder, which exhibits a BET surface area (200 m2 g−1) comparable with that of the titanium silicate carrier applied in the commercial catalyst (Fig. 4). A type IV isotherm is observed characteristic of mesoporous materials with a high uptake at mid-high relative pressures (p/p0 > 0.5) accompanied by type 2 hysteresis. The corresponding pore size distribution derived by the Barrett–Joyner–Halenda (BJH) model applied to the adsorption branch of the isotherm evidences a discrete range of mesopore sizes centered around 10 nm. Comparatively, the monolith catalyst exhibits a significantly reduced surface area and pore volume (Table 1), which is consistent with the low mass fraction of the γ-Al2O3 coating (ca. 3 wt% when considering the large volume of the steel frame). Nonetheless, the equivalent form of the isotherm and size of the mesopores evidenced, confirms the similar textural properties of the washcoat layer to the powder catalyst. Synthetic aluminas are known to exhibit surface acidity, which could influence the interaction with the hybrid nanoparticles. The presence of acid sites of varying strength was confirmed by NH3-TPD (Fig. 3c); a single broad peak could be identified in the desorption profiles reaching a maximum at around 500 K.37 The Lewis acidic nature of the sites was confirmed by the infrared study of adsorbed pyridine.38 Quantitative analysis of the bands at 1623 and 1613 cm−1 evidenced a higher concentration of strong than medium-strength sites (71 and 200 μmol g−1, respectively). However, comparative analysis of the TiSi2O6 carrier used in NanoSelect also evidenced an appreciable concentration of acid sites of similar strength, indicating that the surface properties of the carrier should not significantly affect the performance of the hybrid nanoparticles in the monolith catalyst.
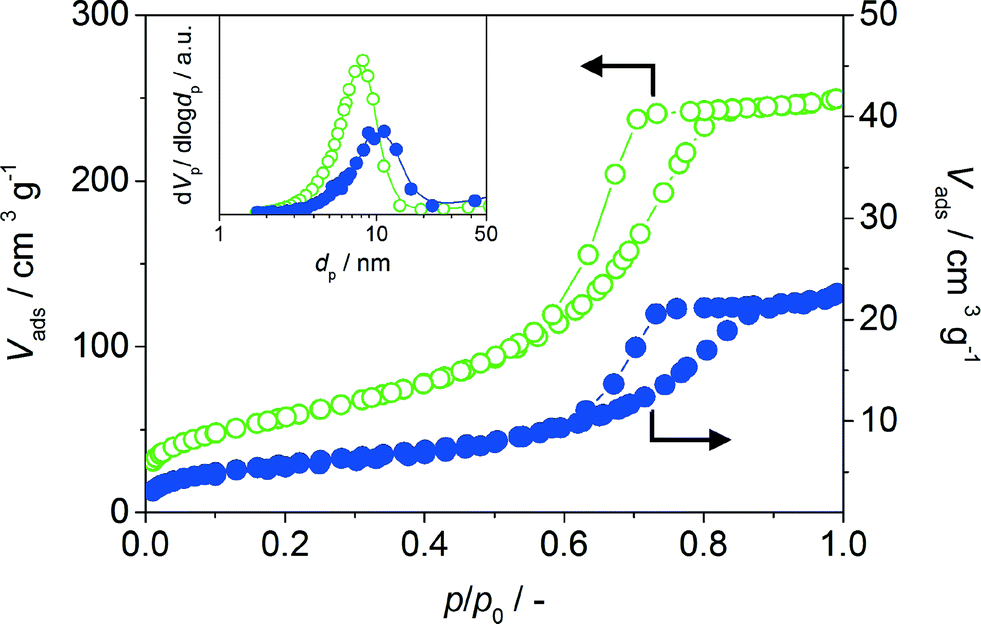 | ||
| Fig. 4 N2 isotherms of Pd-HHDMA/Al2O3-p (open circles) and of Pd-HHDMA/Al2O3-m (solid circles). The inset shows the corresponding BJH pore size distributions derived from the adsorption branch of the isotherm. | ||
No alteration of the crystalline order or of the aluminium speciation was observed upon impregnation of the Pd-HHDMA nanoparticles (Fig. 3a and b). The monolith displayed a slightly higher Pd content than the powder catalyst (Table 1). Nonetheless, the total mass of Pd in the monolithic reactor is comparable with that of the packed bed as the mass of catalyst applied is lower (ca. 1/3) in the former case. Since it is known that the HHDMA ligand can interact with the carrier as well as the Pd, thermogravimetric analysis in air was undertaken to determine the organic content and relative stability of Pd-HHDMA/Al2O3 (Fig. S1†). A very close agreement was observed in the decomposition profile of Pd-HHDMA when supported on Al2O3 or on TiSi2O6, with a gradual weight loss of similar magnitude between 400–530 K associated with the removal of physisorbed water, followed by a sharp weight loss between 550–600 K, assigned to the decomposition of the ligand. Importantly, this indicates that the nature of the carrier does not impact the stability of the active phase. The higher weight loss observed in the second step in the Pd-HHDMA/Al2O3 sample (13 versus 9.6 wt%) could be related to the smaller average particle size of the Pd-HHDMA nanoparticles with respect to the commercial catalyst (5 versus 8 nm) or to differences in the amount of free ligand in the initial Pd/HHDMA colloid.
Monolith architecture
Different techniques have been applied to examine the three dimensional architecture of the monolith catalyst. Imaging by micro-CT enabled the non-destructive visualisation of the whole monolith (Fig. 5a–c). The 19 parallel microchannels are clearly visible in a two dimensional virtual slice (Fig. 5a). The 3D reconstruction of a single image stack (Fig. 5b) evidenced noticeable surface rugosity in the range of tens of microns. Nonetheless, the channels remained unblocked (Fig. 5c) in all cases. Unfortunately, despite applying an advanced edge-contrast enhancement routine, the absorptive and refractive differences between the steel monolith and the comparatively light porous alumina layer were insufficient to distinguish the interface of these phases. In fact, as shown in Fig. S2,† similar protrusions into the microchannel are observed before and after coating, corroborating the complex topography of the metal surface. | ||
| Fig. 5 (a) Two-dimensional virtual slice of a perpendicular cross-section computed through micro-CT. 3D reconstructions derived from the micro-CT data of (b) one quarter of the monolith and (c) a single microchannel within the same section. Back-scattered electron image and corresponding elemental map of aluminium (red) and iron (blue) of (d, e) a longitudinal cross-section of microchannel and (f, g) a lateral cross-section of a single microchannel, illustrating the uniformity of the washcoat layer over the surface of the metallic monolith. (h) HAADF-STEM image and (i) corresponding elemental map of aluminium (red) and palladium (green) confirming the good dispersion of the Pd-HHDMA nanoparticles over the carrier, and (j) a higher magnification HAADF-STEM image of the Pd-HHDMA nanoparticles. | ||
To study the distribution of the washcoat, it was thus necessary to resort to destructive techniques, which was approached by precision sectioning. Back-scattered electron imaging by SEM and correlative elemental maps of iron and aluminium acquired by EDS of longitudinal (Fig. 5d and e) and latitudinal (Fig. 5f and g) cross-sections enabled the clear distinction of the washcoat layer from the steel skeleton. A thin coating of alumina was seen to penetrate along the microchannels, separated by the nonporous steel walls. The average thickness of the washcoat in the center of the monolith varied from a few to ca. 50 μm. This heterogeneity reflected the roughness of the metal surface, which was seen to protrude into the microchannels over a similar size range. The reduced average thickness with respect to that determined at the end of the monolith by optical microscopy, emphasises the importance of characterising the washcoat distribution along the longitudinal axis.
Finally HAADF imaging by STEM and correlative EDS mapping of aluminium (red) and palladium (green) were conducted to access the nanostructural features of the monolithic reactor (Fig. 5h–j). The Pd-HHDMA nanoparticles are observed to decorate the γ-Al2O3 carrier, appearing well distributed across the sample. High magnification images confirm that they preserve a narrow particle size distribution, centred around 5 nm in diameter, which is consistent with the size of the nanoparticles in the colloidal solution. The porous nature of the γ-Al2O3 layer is also visible, comprising a dense network of aggregated nanoparticles of ca. 10–20 nm in size. The packing of these particles gives rise to the mesoporosity evidenced by gas sorption (Fig. 4).
Hydrogenation of alkynes
The performance of the catalysts was first evaluated in the flow hydrogenation of 2-methyl-3-butyn-2-ol to 2-methyl-3-buten-2-ol (Fig. 6a), an important reaction in the synthesis of fragrances, cosmetics, and vitamins. The full reaction scheme and a representative GC chromatogram obtained during the product analysis are reported in Fig. S3 and S4.† The contour plots in Fig. 6a show the influence of temperature (T = 303–363 K) and pressure (P = 1–8 bar) on the conversion of 2-methyl-3-butyn-2-ol and on the selectivity to 2-methyl-3-buten-2-ol over Pd-HHDMA/Al2O3-m. An excellent degree of product selectivity (>90%) is evidenced at almost all of the conditions studied, even at high temperature and pressure (i.e., at T > 343 K and P > 5 bar). This result is particularly interesting if compared with packed-bed reactors based on Pd-HHDMA/Al2O3-p and the Nanoselect catalysts (Fig. 6b and c, respectively), which show a much lower degree of product selectivity at similar conversion levels. For example, at 363 K and 3 bar, Pd-HHDMA/Al2O3-m displays a selectivity to 2-methyl-3-buten-2-ol of 100% at a degree of alkyne conversion of 55%, while Pd-HHDMA/Al2O3-p and NanoSelect display 83% and 81% alkene selectivity at an even lower conversion (ca. 40%). It is noteworthy that, for NanoSelect and Pd-HHDMA/Al2O3-p, the inert carrier (TiSi2O6 or γ-Al2O3, respectively) was homogenously mixed as a diluent to fill the cartridge. The similarity of the catalytic results for a broad range of conditions indicates that the type of carrier do not affect the catalytic performance. The observed higher alkene selectivity in the monolith at elevated temperature and pressure is likely due to the more efficient heat dissipation in a metallic monolith.12 In fact, in a highly-exothermic reaction such as alkyne hydrogenation, heat is released (ΔHalkyne semi-hydrogenation = 40 kJ mol−1). Consequently, temperature gradients can develop within and around the catalyst pellet. The use of stainless steel for the monolith body likely facilitates the removal of heat, avoiding the formation of local hot spots and permitting high selectivity also at higher temperatures and pressures. | ||
| Fig. 6 Conversion (in %, left) and selectivity to 2-methyl-3-buten-2-ol (in %, right) in the hydrogenation of 2-methyl-3-butyn-2-ol over Pd-HHDMA/Al2O3-m (a), NanoSelect (b), Pd-HHDMA/Al2O3-p diluted with Al2O3 (c), and Pd-HHDMA/Al2O3-p diluted with SiC (d). Conditions: Wcat = 0.1 g, FL(2-methyl-3-butyn-2-ol + toluene) = 1 cm3 min−1, FG(H2) = 36 cm3 min−1, τmonolith = 4.2 s−1, τpacked-bed = 4.7 s−1. Each contour plot was obtained through the spline interpolation of the 16 experimental points indicated with dots. | ||
To quantify the advantage of heat dissipation, we have compared the thermal conductivity of the monolith with the one of packed-bed reactor by calculating the ratio between the heat transfer coefficient and the thermal conductivity of the solid, following the approach reported by Flytzani-Stephanopoulos and co-workers.39 A low ratio is expected to result in a good heat dissipation, leading to a higher selectivity to the desired product. As expected, the ratio calculated for a stainless steel monolith is much lower than that for an Al2O3-supported bed (40 versus 60, respectively). To corroborate this hypothesis, we have conducted an additional experiment in which the cartridge used for the catalytic tests with Pd-HHDMA/Al2O3-p was packed with silicon carbide rather than γ-Al2O3 as a diluent (Fig. 6d). Silicon carbide is a ceramic material characterised by a low thermal expansion coefficient, good thermal shock resistance, and excellent thermal conductivity (120 W m−1 K−1), much higher than those of alumina or titanium silicate (18 and 20 W m−1 K−1, respectively). Also in this case, due to the improved heat transfer, an outstanding alkene selectivity was observed in a broad range of conditions (i.e., above 343 K and 5 bar) and the results paralleled those of the monolith.
Another advantage of the monolithic reactor is its relatively low pressure drop. Although we cannot directly measure this variable due to the small average free length of the channels in the monolith (t = 0.4 mm), we have applied the Lockhart–Martinelli correlation to estimate the extent of overall pressure drops in the reactors.40 Though the values are small in both cases, the monolith is expected to experience a lower overall pressure drop (0.1 mbar) compared to the packed-bed reactor (4 mbar). Based on the negligible values, we do not believe that the observed heterogeneity in the washcoat coating will result in appreciable differences in pressure drop (and consequently a distribution of residence times) between the different channels. In fact, although the washcoat thickness varies between 0–50 μm, our observations indicate that the heterogeneity is similar in all of the channels, therefore no significant difference can be expected. This is also in line with earlier papers highlighting flow maldistribution leading to pressure drop variations can be neglected for monoliths with channel diameter lower than 0.1 mm.41
Given that a major drawback of structured reactors is the low resistance towards leaching of the active phase, a stability test was conducted at 303 K and 3 bar over Pd-HHDMA/Al2O3-m. As shown in Fig. 7, no drop in activity and selectivity was observed for more than 20 h, confirming the secure anchoring of the hybrid nanoparticles in the monolith via ligand-support interactions. This is consistent with the strong dispersion interactions previously evidenced between the orthophosphoric head of the ligand and the titanium silicate and activated carbon carriers, which emerge even at low HHDMA contents and increase with the surface ligand concentration.6,7 Unfortunately, it was not possible to directly compare the Pd content in the as-prepared and the used monolith due to the destructive nature of compositional analysis by ICP-OES. However, we have measured the Pd content in three different monoliths, which were freshly prepared or used in hydrogenations. The variation observed was within 5%, further indicating the effective stabilisation of the nanoparticles on the support.
 | ||
| Fig. 7 Stability test over the monolith Pd-HHDMA/Al2O3-m in the hydrogenation of 1-hexyne to 1-hexene. Conditions: T = 303 K, P = 3 bar, FL(1-hexyne + toluene) = 1 cm3 min−1, FG(H2) = 36 cm3 min−1, τmonolith = 4.2 s−1. | ||
Finally, the results for 2-methyl-3-butyn-2-ol were extrapolated to other alkynes with internal or external triple bonds and similar functionality (3-hexyne, 4-octyne, 3-methyl-1-pentyn-3-ol; see Fig. 8). In all cases, the monolithic reactor provided a better performance compared to conventional powdered materials, corroborating the possibility to effectively structure hybrid Pd nanoparticles in monoliths for applications in the selective hydrogenation of a broad range of acetylenic compounds.
 | ||
| Fig. 8 Selectivity to cis-3-hexene, cis-4-octane, and 3-methyl-1-penten-3-ol in the hydrogenation of different acetylenic substrates over NanoSelect (black), Pd-HHDMA/Al2O3-p diluted with Al2O3 (red), Pd-HHDMA/Al2O3-p diluted SiC (blue), and Pd-HHDMA/Al2O3-m (green). Conditions: T = 343 K, P = 5 bar FL(alkyne + toluene) = 1 cm3 min−1, FG(H2) = 36 cm3 min−1, τmonolith = 4.2 s−1, τpacked-bed = 4.7 s−1. The conversion level was between 50–70% in all cases. | ||
Conclusions
We have prepared and characterised metallic monolithic reactors featuring hybrid HHDMA-modified Pd nanoparticles. The synthesis involved the controlled deposition of a boehmite washcoat on the metallic walls of the monolith, the hardening to form a well-adhered porous gamma alumina layer, and the subsequent immobilisation of Pd-HHDMA. Dedicated microscopy and tomography techniques were essential to gain insight into the structural uniformity of the steel skeleton and washcoat layer and the high dispersion of the active phase. Evaluation of the material in the continuous-flow three-phase hydrogenation of functionalised acetylenic compounds confirmed the high resistance to leaching of the ligand-modified Pd. Advantageously, when stabilised in the monolithic reactor, the hybrid nanoparticles preserved the intrinsic selective character over a wider range of operating conditions. This was attributed to the high thermal conductivity of the metallic skeleton of monolith, which helps removing the heat generated during reaction and results in an enhanced isothermicity of the catalyst bed. The selectivity trends evidenced over packed-bed reactors diluted with high-thermal conductivity silicon carbide support this conclusion. Overall, this study provides a unified approach to drive catalyst design from the nanoscale (controlled surface composition in hybrid Pd-HHDMA nanoparticles) to the reactor level (incorporation of hybrid nanoparticles in monolithic body), in order to obtain catalytic materials with superior performance in flow hydrogenation.Acknowledgements
Financial support from ETH Zurich is acknowledged. Dr Peter T. Witte is thanked for providing the colloidal solution of Pd-HHDMA. Michael Ehrenstein and Dr René Verel are thanked for help with the monolith preparation and MAS NMR measurements, respectively. The Scientific Centre for Optical and Electron Microscopy (ScopeM) of ETH Zurich is acknowledged for providing access to its facilities.References
- C. Sanchez, K. J. Shea and S. Kitagawa, Chem. Soc. Rev., 2011, 40, 471 RSC.
- G. E. Oosterom, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2001, 40, 1828 CrossRef CAS.
- A. Kaushik, R. Kumar, J. K. Arya, M. Nair, B. D. Malhotra and S. Bhansali, Chem. Rev., 2015, 115, 4571 CrossRef CAS PubMed.
- P. T. Witte, P. H. Berben, S. Bolan, E. H. Boymans, D. Vogt, J. W. Geus and J. G. Donkervoort, Top. Catal., 2012, 55, 505 CrossRef CAS.
- P. T. Witte, S. Boland, F. Kirby, R. van Maanen, B. F. Bleeker, D. A. M. de Winter, J. A. Post, J. W. Geus and P. H. Berben, ChemCatChem, 2013, 5, 582 CrossRef CAS.
- G. Vilé, N. Almora-Barrios, S. Mitchell, N. López and J. Pérez-Ramírez, Chem. – Eur. J., 2014, 20, 5926 CrossRef PubMed.
- G. Vilé, N. Almora-Barrios, N. López and J. Pérez-Ramírez, ACS Catal., 2015, 5, 3767 CrossRef.
- D. Albani, G. Vilé, S. Mitchell, P. T. Witte, N. Almora-Barrios, R. Verel, N. López and J. Pérez-Ramírez, Catal. Sci. Technol., 2016, 6, 1621 CAS.
- G. Vilé, D. Albani, N. Almora-Barrios, N. López and J. Pérez-Ramírez, ChemCatChem, 2016, 8, 21 CrossRef.
- A. Sachse, A. Galarneau, B. Coq and F. Fajula, New J. Chem., 2011, 35, 259 RSC.
- T. A. Nijhuis, A. E. W. Beers, T. Vergunst, I. Hoek, F. Kapteijn and J. A. Moulijn, Catal. Rev.: Sci. Eng., 2001, 43, 345 CAS.
- A. Renken and L. Kiwi-Minsker in Advance in Catalysis, ed. B. C. Gates and H. Knözinger, Elsevier, 2010, vol. 53, pp. 47–121 Search PubMed.
- L. Gini, C. Cristiani, G. Groppi and E. Tronconi, Appl. Catal., B, 2006, 62, 121 CrossRef.
- A. F. Pérez-Cadenas, M. M. P. Zieverink, F. Kapteijn and J. A. Moulijn, Catal. Today, 2005, 105, 623 CrossRef.
- A. Sachse, N. Linares, P. Barbaro, F. Fajula and A. Galarneau, Dalton Trans., 2013, 42, 1378 RSC.
- P. Pfeifer, A. Kölbl and K. Schubert, Catal. Today, 2005, 110, 76 CrossRef CAS.
- N. Semagina, M. Grasemann, N. Xanthopoulos, A. Renken and L. Kiwi-Minsker, J. Catal., 2007, 251, 213 CrossRef CAS.
- N. Linares, S. Hartmann, A. Galarneau and P. Barbaro, ACS Catal., 2012, 2, 2194 CrossRef CAS.
- Y. Elias, P. R. von Rohr, W. Bonrath, J. Medlock and A. Buss, Chem. Eng. Process., 2015, 95, 175 CrossRef CAS.
- E. V. Rebrov, E. A. Klinger, A. Berenguer-Murcia, E. M. Sulman and J. C. Shouten, Org. Process Res. Dev., 2009, 13, 991 CrossRef CAS.
- J. Kobayashi, Y. Mori, K. Okamoto, R. Akiyama, M. Ueno, T. Kitamori and S. Kobayashi, Science, 2004, 304, 1305 CrossRef CAS PubMed.
- N. Wang, T. Matsumoto, M. Ueno, H. Miyamura and S. Kobayashi, Angew. Chem., Int. Ed., 2009, 48, 4744 CrossRef CAS PubMed.
- A. Gömann, J. A. Deverell, K. F. Munting, R. C. Jones, T. Rodemann, A. J. Canty, J. A. Smith and R. M. Guijt, Tetrahedron, 2009, 65, 1450 CrossRef.
- F. Costantini, E. M. Benetti, R. M. Tiggelaar, H. J. G. E. Gardeniers, D. N. Reinhoudt, J. Huskens, G. J. Vancso and W. Verboom, Chem. – Eur. J., 2010, 16, 12406 CrossRef CAS PubMed.
- U. Kunz, A. Kirschning, H.-L. Wen, W. Solodenko, R. Cecilia, C. O. Kappe and T. Turek, Catal. Today, 2005, 105, 318 CrossRef CAS.
- S. Ungureanu, H. Deleuze, C. Sanchez, M. I. Popa and R. Backov, Chem. Mater., 2008, 20, 6494 CrossRef CAS.
- F. Liguori and P. Barbaro, J. Catal., 2014, 311, 212 CrossRef CAS.
- D. Paganin, S. C. Mayo, T. E. Gureyev, P. R. Miller and S. W. Wilkins, J. Microsc., 2002, 206, 33 CrossRef CAS PubMed.
- G. R. Myers, S. C. Mayo, T. E. Gureyev, D. M. Paganin and S. W. Wilkins, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 76, 045804 CrossRef.
- M. Valentini, G. Groppi, C. Cristiani, M. Levi, E. Tronconi and P. Forzatti, Catal. Today, 2001, 69, 307 CrossRef CAS.
- G. F. Froment and K. B. Bischoff, Chemical reactor analysis and design, Wiley, 1990, p. 335 Search PubMed.
- W. Liu, AIChE J., 2002, 48, 1519 CrossRef CAS.
- A. K. Heibel, P. J. M. Lebens, J. W. Middelhoff, F. Kapteijn and J. Moulijn, AIChE J., 2005, 51, 122 CrossRef CAS.
- F. H. M. Dekker, A. Bliek, F. Kapteijn and J. A. Moulijn, Chem. Eng. Sci., 1995, 50, 3573 CrossRef CAS.
- T. A. Nijhuis, G. Van Koten, F. Kapteijn and J. A. Moulijn, Catal. Today, 2003, 79, 315 CrossRef.
- R. H. Meinhold, R. C. T. Slade and R. H. Newman, Appl. Magn. Reson., 1993, 4, 121 CrossRef CAS.
- S.-M. Kim, Y.-J. Lee, J. W. Bae, H. S. Potdar and K.-W. Jun, Appl. Catal., A, 2008, 348, 113 CrossRef CAS.
- M. R. Gafurov, I. N. Mukhambetov, B. V. Yavkin, G. V. Mamin, A. A. Lamberov and S. B. Orlinskii, J. Phys. Chem. C, 2015, 119, 27410 CAS.
- M. Flytzani-Stephanopoulos, G. E. Voecks and T. Charng, Chem. Eng. Sci., 1986, 41, 1203 CrossRef CAS.
- C. Satterfield, AIChE J., 1975, 21, 209 CrossRef CAS.
- A. Rouge, B. Spoetzl, K. Gebauer, R. Schenk and A. Renken, Chem. Eng. Sci., 2001, 56, 1419 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterisation and kinetic data. See DOI: 10.1039/c6re00114a |
| This journal is © The Royal Society of Chemistry 2016 |