
Continuous flow synthesis concatenated with continuous flow liquid–liquid extraction for work-up and purification: selective mono- and di-iodination of the imidazole backbone†
Audun
Drageset
and
Hans-René
Bjørsvik
*
Department of Chemistry, University of Bergen, Allégaten 41, N-5007 Bergen, Norway. E-mail: hans.bjorsvik@kj.uib.no; Web: http://folk.uib.no/nkjhn/
First published on 5th July 2016
Abstract
Flow processes for mono- and di-iodination of the imidazole backbone were devised, developed, and implemented on the multi-jet oscillating disk (MJOD) flow reactor platform. The flow processes were based on batch protocols previously developed in our research group and involved N,N′-1,3-diiodo-5,5-dimethylhydantoin as the iodination reagent. The flow processes demanded short reactor residence times, and afforded the mono- or di-iodinated imidazoles at a high mass × time yield [g min−1]. The continuous flow processes leading to 4(5)-iodo-1H-imidazole HCl salt was concatenated with a continuous flow work-up section that was assembled by a series of hold-up tanks and two additional MJOD flow reactors that both were utilized as liquid–liquid extractors. The overall mass throughput allowed a production of 4(5)-iodo-1H-imidazole HCl salt at a capacity of 95 g day−1. The continuous flow reaction unit utilized for the preparation of 4,5-diiodo-1H-imidazole was concatenated with a double set of hold-up tanks/stirred tank-batch reactors and semi-continuous vacuum filtration units for the isolation of precipitated/crystallized 4,5-diiodo-1H-imidazole at a production capacity of 295 g day−1.
Introduction
A long-lasting project in our research group concerns the design and development of synthetic methodology for the functionalization of the imidazole framework in general and the imidazole backbone in particular. The backbone elaboration of the imidazole ring (Scheme 1) commences with iodination of imidazole 1 (ref. 1) to provide 4,5-diiodo-1H-imidazole 2 [path (a)] or 4(5)-iodo-1H-imidazole 3 by selective iodination on position-4 [path (c)] or via a di-iodination combined with a hydrodeiodination sequence,1c,2a paths (a + b). | ||
| Scheme 1 The elaboration of the imidazole backbone commences with iodination using N,N′-diiodo-5,5-dimethylhydantoin (DIH) (pathway a or b), that is followed by installation of a N-auxiliary group (AUX) (pathway d or e). The ultimate step comprises a coupling reaction; Suzuki cross-coupling2a (path f), Stille coupling2b (path g), or Sonogashira coupling2c (path h). | ||
The two backbone iodinated imidazoles 2 and 3, emerge as key intermediates for the synthesis of several backbone-modified imidazoles. In this context, we devised and developed small-scale laboratory protocols for the iodination of the imidazole backbone.1f Both the mono- and diiodo-imidazoles were in a following step furnished with a N-auxilliary group; tosyl (Tos) or N,N-dimethylaminosulfonyl (DMAS), which we have observed to be compulsory in several cases when subsequent elaboration of the imidazole backbone was conducted.2
A medicinal chemistry project in progress in our group required multi-gram supplies of 4(5)-iodo-1-tosyl-1H-imidazole 5a, which is an intermediate in the synthesis of backbone substituted Ag–NHC complexes revealed to exhibit cytotoxic effect in vitro in leukemia cells.3 Further in vivo animal studies were planned, which requested a scaled-up process that could supply the intermediates and ultimately the active substance in a reliable and timely fashion. For this purpose, our small-scale laboratory procedure1f for the iodination of the imidazole backbone appeared attractive for scale-up due to its high selectivity and reaction rate.
However, the small-scale batch procedure based on the labile iodination reagent DIH in sulfuric acid is challenging on a large-scale, because a scaled-up procedure continuously demands small batches of freshly prepared reagent.
Transfer of the small-batch synthesis into a continuous flow process appeared as a more appropriate solution.4 We have previously established a multi-100 gram scale DIH process5 that furnishes the iodination agent. This process comprises a flow synthesis step using our multi-jet oscillating disk (MJOD) continuous flow reactor platform6 telescoped with a semi-continuous product isolation step using vacuum filtration.
Results and discussion
Flow process leading to 4,5-diiodo-1H-imidazole (2)
At the outset of this study, we encountered a mostly unsolved problem in contemporary flow chemistry; how to prepare and feed a homogeneous slurry to a flow milli/micro-reactor. Although, some few flow chemistry applications involving slurry-forming reaction have been disclosed; Ley and collaborators7 investigated an agitated cell reactor, our recently disclosed DIH process,5 and some few more examples are summarized in a review dedicated to upstream continuous processing of fine chemicals.8 In general, a flow micro-reactor cannot be used for reactions involving slurries, as the narrow channels and tubing will be clogged by the solid slurry particles, while our in-house developed MJOD flow reactor platform has been successfully proven to handle reaction mixtures where solids precipitate during the course of the reaction.5,6 From an environmental, economical, and chemical compatibilities point of view, water appeared as an appropriate reaction medium for the flow iodination process. However, DIH shows low solubility in water and forms thus a slurry with water. Nevertheless, attempts to directly feed the slurry appeared not viable due to clogging of the tubing that interconnected the reagent reservoir (R1) and the feeding pump (P1) after only short working time. The feeding pump P1 was furnished with a valve-less ceramic piston pump head appropriate for pumping slurries.9 This pump operates with a low piston stroke rate (0.30 s−1). This resulted in accumulation of solid particles on the tube wall that finally clogged the reagent feeding system. In an attempt to overcome this problem in the feeding tubing circuit, we attempted a previously disclosed method;10 N2 gas was inserted via valve V1 in order to create a plug flow by alternating gas and slurry. In addition, a valve, V2, was inserted at a position close to the pump (P1) inlet, see Fig. 1. | ||
| Fig. 1 (A) A small section of the flow chart for the continuous flow iodination process where a plug flow arrangement was incorporated in an effort to achieve a homogeneous feeding of a DIH – water slurry. (B) (a) Normal operation of the slurry feeding by means of a plug flow by alternating gas and slurry. (b) The pipeline is clogged, and the feeding is terminated. (c) The solid plug is removed by closing valve V1 and open valve V2 that furnish a slightly elevated pressure of N2 that brush the solid plug back into the reservoir R1. | ||
This gas inlet valve (V2) was inserted in the flow circuit as a tool devoted to remove “solid plugs” that precipitated inside the tubing-interconnecting reservoir R1 and the pump P1. By exposing the tubing system with a slightly elevated pressure via valve V2, precipitated solids could be pushed back into the reservoir R1, whereupon the slurry feeding from R1 once again could be started. An outline for this arrangement is given in Fig. 1B. The outlined setup for plug flow lowered the clogging rate with a stop frequency to ≈ 0.13 incidents min−1. However, the outlined process required continuous monitoring and supervision by an operator, which made this method very laborious and was thus abandoned.
In an attempt to resolve the clogging problem DIH was mixed with sulfuric acid in the reagent reservoir, an alteration that afforded target compound in moderate-to-good yield. The continuous flow reactor-setup as shown in Fig. 2, section (a) was used in to order to operate this process. Multiple freshly prepared mixtures of DIH and H2SO4 was prepared during the process run in order to minimize decomposition of the DIH reagent. Overall, the approach materialized thus to be inappropriate for up scaling due to challenges in the practical performance. A third method we explored involved standard reagent feeding of a solution (DIH in methanol). The ultimate method was proved to most reliable and was used for the further experimentation.
 | ||
| Fig. 2 Flow diagram for the flow process leading to 4,5-diiodo-1H-imidazole 2. The entire process is divided in three various sections. Section (a) is constituted by a selective flow di-iodination of the 4- and 5-position of the imidazole backbone and reaction quenching using a MJOD flow reactor (MJOD1). Section (b) is constituted of the post-reaction mixture collection in a hold-up tank R5a,b-STR whereupon a pH regulation is conducted. Section (c) is a semi-continuous filtration using vacuum filter-nutches F1a and F1b. | ||
The multi-jet oscillating disk (MJOD) flow reactor rig [MJOD1, section (a) of Fig. 2] was used to conduct the iodination in continuous flow. The rig was equipped with several sets of reservoirs and pumps; R1 containing imidazole 1 dissolved in a mixture of water and sulfuric acid mixture. DIH dissolved in methanol was placed in reservoir R2. Details about the design and operation of the MJOD continuous flow reactor have previously been reported,6 but technical specifications for the MJOD reactor set-up used herein are provided in Table 2.
The MJOD flow reactor (MJOD1) was adjusted and regulated at a temperature of 0 °C by means of a cooler (C/HE1) and the reagents were pumped (P1 and P2) into the MJOD flow reactor at rates that afforded a reactor residence time of 15 min.
The reaction mixture was quenched inline by pumping (P3) aqueous sodium hydroxide from reservoir R3 into the MJOD flow reactor at a short distance before the reactor outlet. The quenched reaction mixture was collected in a hold-up tank (R5a-STR). When this reservoir was nearly full, the reactor output flow was diverted to the empty reservoir (R5b-STR).
Batch mode neutralization (R5a,b-STR) was then performed by adding glacial acetic acid from reservoir R4 whereupon target product 4,5-diiodo-1H-imidazole precipitated, section (b) of Fig. 2. The precipitated/crystallized target product 2 was filtered-off in a semi-continuous fashion (alternating between the filter nutches F1a and F1b), see section (c) of Fig. 2. The overall process leading to target product 4,5-diiodo-1H-imidazole 2 proceeded with a practically quantitative yield. A 30 min. Test run including imidazole 1 (1.323 g, 19.4 mmol) and DIH (12 g, 31.6 mmol) provided a yield of ≈97% of target 4,5-diiodo-1H-imidazole 2 (6.02 g, 18.8 mmol).
Flow process leading to 4(5)-iodo-1H-imidazole (3)
An analogue mono-iodination process using DIH was attempted established using an increased dilution and an adjusted ratio between quantities of the DIH reagent versus the substrate imidazole 1. However, the mono-iodination did not operate as anticipated and afforded target compound 4(5)-iodo-1H-imidazole HCl salt in low yields only (9–13%).The mono-iodination process was thus subjected for scale-up and optimization by means of statistical experimental design11 and multivariate modeling using multiple linear regression.12 Furthermore, the design of the continuous flow process was planned to be a 10× scale-up of the corresponding batch procedure. Prior to the experimental investigation, a cause–effect analysis of the process was performed and summarized in the Ishikawa cause–effect diagram13 of Fig. 3.
 | ||
| Fig. 3 Ishikawa cause–effect diagram for the iodination processes. | ||
The reaction was explored using a fractional factorial design of type 2k−1 + c, k = 4, c = 3, with resolution R = IV,14 which provides a design matrix D that includes the experiments #1–11 of Table 1. The experimental space was spanned by the variables z1, …, z4 and a model matrix M was created using eqn (1). The MATLAB15 software was used for the calculations and graphical presentations of adjacent line graphics.
| Exp. levels | ||||||
|---|---|---|---|---|---|---|
| Exp. variables | Unit | −1 | 0 | +1 | ||
a Generator: x4 = x1 × x2 × x3 that provides a resolution R = IV design with the alias pattern of the regression coefficients.
b Responses: y1 = isolated yield% of 4(5)-iodo-1H-imidazole HCl salt, y2 = isolated yield% of 4,5-diiodo-1H-imidazole.
c The experiment #1, 8, and 9 were performed first in order to assure a sufficient span in results in the extreme points of the experimental domain. Thereafter the rest of the design was conducted in a random order.
d Calculated statistics for objects #9, 10, and 11: ![[y with combining overline]](https://www.rsc.org/images/entities/char_0079_0305.gif) 1 = 32.733 and s1 = 0.6506. 1 = 32.733 and s1 = 0.6506.
|
||||||
| z 1 | Reaction time | [min] | 5 | 10 | 15 | |
| z 2 | Solvent volume | [mL] | 80 | 110 | 140 | |
| z 3 | Quantity DIH | [equiv.] | 0.4 | 0.5 | 0.6 | |
| z 4 | Acid volume | [mL] | 3 | 6 | 9 | |
| Statistical experimental design | ||||||
|---|---|---|---|---|---|---|
| Entry | Exp. variables | Responsesb | ||||
| z 1 | z 2 | z 3 | z 4 | y 1 | y 2 | |
| 1c | 5 | 80 | 0.4 | 3 | 37.0 | 0.8 |
| 2 | 15 | 80 | 0.4 | 9 | 52.0 | 0.0 |
| 3 | 5 | 140 | 0.4 | 9 | 22.6 | 5.0 |
| 4 | 15 | 140 | 0.4 | 3 | 44.0 | 0.0 |
| 5 | 5 | 80 | 0.6 | 9 | 24.8 | 0.0 |
| 6 | 15 | 80 | 0.6 | 3 | 35.0 | 0.0 |
| 7 | 5 | 140 | 0.6 | 3 | 23.0 | 0.0 |
| 8c | 15 | 140 | 0.6 | 9 | 38.6 | 4.4 |
| 9c,d | 10 | 110 | 0.5 | 6 | 32.7 | 8.2 |
| 10d | 10 | 110 | 0.5 | 6 | 33.4 | 10.7 |
| 11d | 10 | 110 | 0.5 | 6 | 32.1 | 12.2 |
| Optimization experiments | ||||||
|---|---|---|---|---|---|---|
| Entry | Exp. variables | Responsesb | ||||
| z 1 | z 2 | z 3 | z 4 | y 1 | y 2 | |
| 12 | 20 | 50 | 0.4 | 3 | 57.5 | 0.0 |
| 13 | 25 | 20 | 0.4 | 3 | 63.6 | 0.0 |
| 14 | 30 | 20 | 0.4 | 3 | 65.3 | <0.05 |
The design values (zk) were scaled according to eqn (2) to facilitate the regression analysis and the interpretation of the final empirical model. The experiments that constitutes the design matrix D and responses are shown in Table 1.
The derived empirical model y1 = f(x1, x2, x3, x4) that describes the response yield% of 4(5)-iodo-1H-imidazole 3 HCl salt as a function of the four experimental variables x1, … , x4 is displayed in Fig. 4 as a cumulative normal distribution (CND) plot16 (on the left hand side) and a stem plot of the regression coefficients (at the right hand side in the figure).
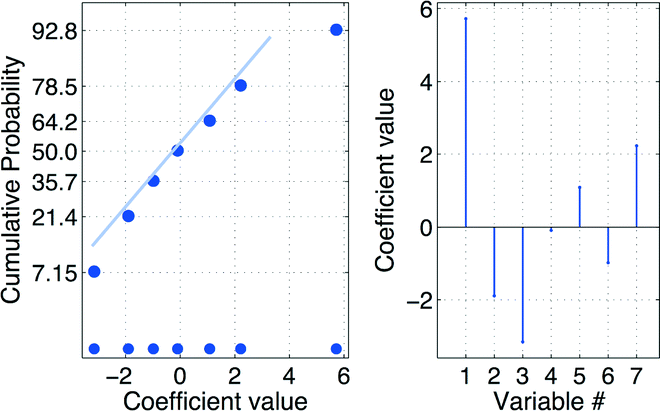 | ||
| Fig. 4 Cumulative normal probability (CND) plot – analysis of the model terms of the estimated model: y = f(x1, x2, x3, x4) = 34.109 + 7.775x1 – 2.575x2 − 4.275x3 – 0.125x4 − 1.475x1x2 (x3x4) − 1.325x1x3 (x2x4) + 3.025x1x4 (x2x3), where the terms given in brackets (xnxm) is the model term alias. | ||

| (1) |

| (2) |
| b = (MTM)−1MTy | (3) |
The two plots suggest that the model parameters for the experimental variables x1, x2, x3 and the two-variable interaction x2x3 were significant, see eqn (4). The estimated model parameters are given in Table 2.
| y = f(x1, x2, x3) = β0 + β1x1 + β2x2 + β3x3 + β23x2x3 | (4) |
| Model terms estimated using: | |||
|---|---|---|---|
| Obj.#1–11 | Obj.#1–12 | Obj.#1–13 | |
| β 0 | 34.109 | 33.854 | 33.825 |
| β 1 | 7.775 | 7.072 | 6.570 |
| β 2 | −2.575 | −1.872 | −1.370 |
| β 3 | −4.275 | −3.924 | −3.885 |
| β 23 | 3.025 | 2.322 | 1.820 |
| R 2 | 0.949 | 0.952 | 0.956 |
| R 2Adj | 0.916 | 0.925 | 0.933 |
| RMSEP | 1.912 | 2.267 | 2.613 |
| RSD | 2.114 | 2.484 | 2.840 |
In short, the model says that a prolonged reaction time is beneficial, which should be combined with shortened solvent volume (more concentrated) and performing the reaction with a deficient quantity of the iodination reagent DIH. Based upon these model predictions, we performed the optimization experiment #12–14. The observed and predicted results are presented in the bar graph of Fig. 5. The prediction that belong to conditions for #12 was performed with the model of eqn (4), the prediction for #13 was predicted with a model that included the achieved experimental results for the experiment #12, and finally the prediction for the conditions of #14 was predicted based on a model that included the achieved results of the #12–13 experiments, see estimated models performed in Table 2. The predicted and observed results for the optimization experiments #12–14 are graphically represented in Fig. 5.
 | ||
| Fig. 5 Predicted by means of eqn (4) and successively updated model as the optimization experiments were conducted. | ||
During the previous batch process development, a reaction temperature of 0 °C was required in order to approach a reasonable good yield.1 Since the reaction temperature was leaved out as an experimental variable in the present statistical experimental design, we decided to perform a subsequent exploration in order to determine whether the reaction temperature is a critical control variable in the flow synthesis. Fig. 6 shows the results form experiments using the optimized process conditions, although performed at four different reaction temperatures, namely −5, 0, 5 and 10 °C. We set-out with an increased temperature of 5 °C, which proved to be highly detrimental for the process, showing a decrease in the process output of 28%-points (43%-relative decrease), and even more at a reaction temperature of 10 °C, namely 41%-point (63%-relative decrease). This shows that the reaction temperature is a variable of paramount importance to control at given value (0 °C). These results spurred us to explore the reaction at lower temperature (−5 °C), which was not beneficial for the outcome.
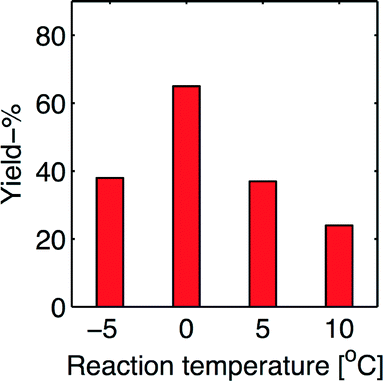 | ||
| Fig. 6 Temperature effect on the optimized procedure. | ||
Liquid–liquid extraction in continuous flow
The isolation of target molecule from the post-reaction mixture was performed by means of liquid–liquid extraction using diethyl ether and water. The extraction process (batch mode) was demanding and time consuming, requested multiple extractions, at least 5× using organic solvent: water in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, to obtain an satisfactory target product recovery.
:1, to obtain an satisfactory target product recovery.
It was therefore desirable to simplify the process and develop a more efficient work-up step. Previously, several studies involving in-line liquid–liquid extraction have successfully been demonstrated.17 Thus we wondered whether the liquid–liquid extraction could be performed with our MJOD continuous flow reactor platform as the extractor unit, a hitherto unexplored application for the MJOD rig.
Each of the work-up steps were first explored as separate operations, see the sections (b)–(d) of Fig. 7. The flow reaction that was conducted in MJOD1 was concatenated with pH regulation (pH 7) in the hold-up tank (R5a,b-STR), section (b) of Fig. 7. The pH regulated post-reaction mixture was then saturated with sodium chloride (S1) and pumped (P5) into the MJOD2 unit and extracted with diethyl ether (P6 and R6). The two-phase output from the extractor MJOD2 was collected in a liquid separator (LS1-R7), where the aqueous phase was drained-off and the organic layer was further processed in a following purification step, section (c) of Fig. 7. This organic phase that contains small quantities of 5,5-dimethylhydantoin was pumped (P7) into the extractor MJOD3. The liquid–liquid extraction was performed with diluted hydrochloric acid (P8 and R8) whereupon a final phase separation was performed in a liquid phase separator tank (LS2-R9). Here the organic phase was drained-off for solvent recovery (R9) and the aquatic phase was pumped (P9) into a rotary evaporator (Evap.), where target product 4(5)-iodo-1H-imidazole hydrochloride was isolated as a pure solid product.
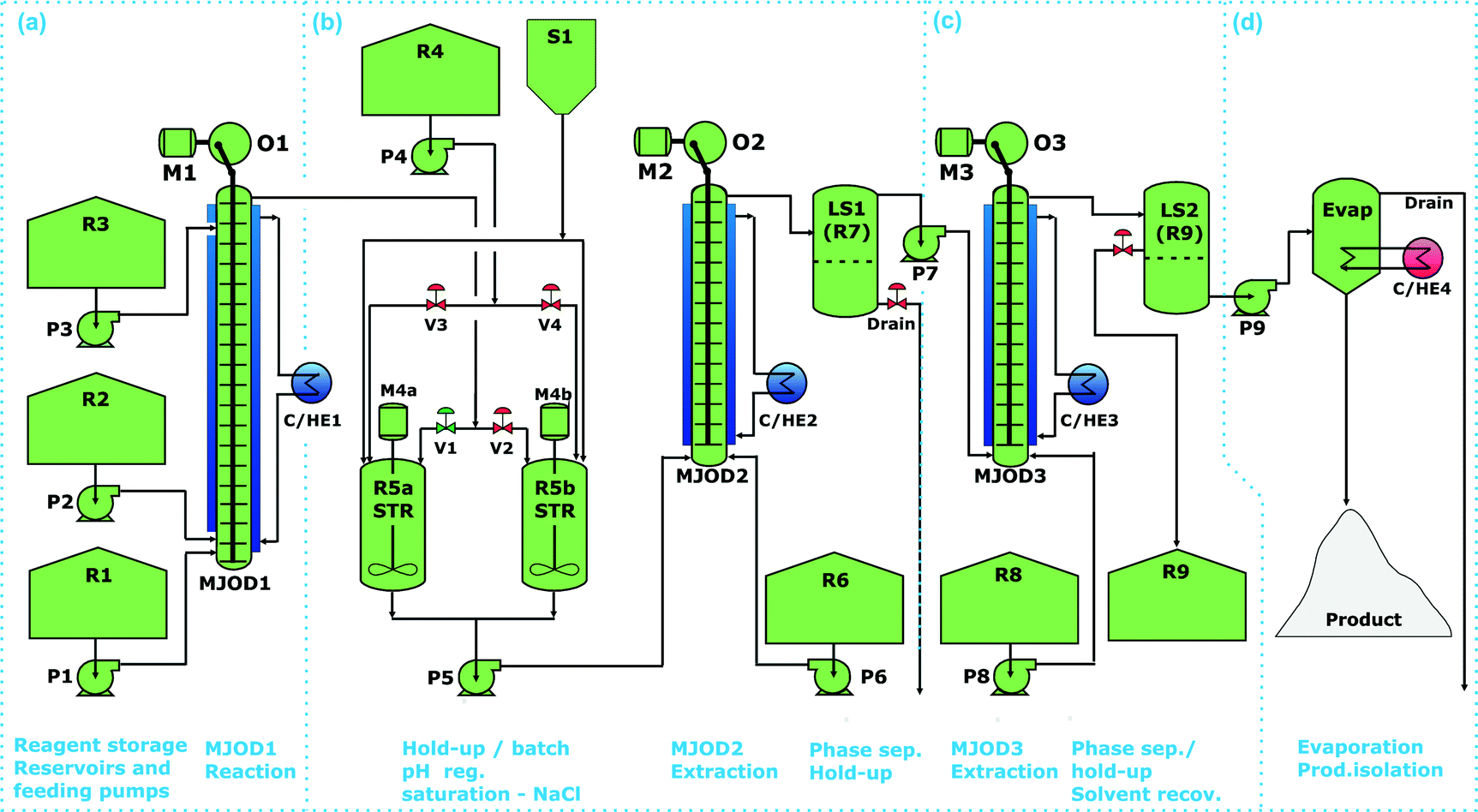 | ||
| Fig. 7 Flow diagram for the flow process leading to 4(5)-iodo-1H-imidazole 3 as a HCl salt. The entire process is divided in four various sections. Section (a) is constituted by the continuous flow selective mono-iodination of the 4-position of the imidazole backbone and reaction quenching. Section (b) collection of post-reaction mixture in a hold-up tank where an pH regulation and saturation also take place. This is connected to a continuous flow liquid–liquid extraction unit that is a MJOD reactor rig connected to a hold-up tank where phase separation (liquid–liquid separation) takes place. Section (c) is a similar operation as outlined in section (b). In section (d), the organic phase from the hold-up/phase separator is then pumped into a rotary evaporator, section (d), where target molecule [4(5)-iodo-1H-imidazole 3 HCl salt] is isolated as a solid. | ||
The various steps (b)–(d) of Fig. 7, was successively attached to the appropriate position as soon as a step was established, developed and successfully tested to ultimately approach the overall continuous flow process as outlined in Fig. 7.
Experimental
General methods
GC-MS analyses were conducted with a capillary gas chromatograph furnished with a fused silica column (L 25 m, 0.20 mm i.d., 0.33 μm film thickness) using a helium pressure of 200 kPa, splitless/split injector and flame ionization detector. DART-MS spectra were obtained with PEG as an internal standard with positive ionization mode with a TOF mass analyzer. 1H NMR spectra were recorded on NMR spectrometers that operating at 400 and 500 MHz and 13C NMR spectra were recorded with NMR spectrometers operating at 100 and 125 MHz. Chemical shifts [ppm] were referenced to the deuterated solvent used for the NMR experiment.The MJOD flow reactor
Previously we have disclosed a detailed report related to the design, construction, and a description for how the multi-jet oscillating disk (MJOD) flow reactor platform works so only a brief account is provided herein. A flow chart with design measurements of the MJOD flow reactors utilized for the iodination processes and the liquid–liquid extraction steps for the product work-up are given in Table 3. The MJOD flow reactor is composed of a short length (height, H) tubular reactor placed in vertical direction. A multi-jet unit is placed in the center of the reactor tube. This MJOD unit is moved by a motive power delivered from a cam wheel that provide an up and down motion (an oscillation) that is driven by a DC motor. Furthermore, the multi-jet unit is composed of a certain number, N, of multi-jet disks and disk spacers (N − 1).| Reaction | Extraction | |||||
|---|---|---|---|---|---|---|
| Continuous flow reactor | Unit | MJOD1 | MJOD2 | MJOD3 | ||

|
Oscillator amplitude range | A | mm | 20 | 20 | 20 |
| Oscillating frequency range | f | Hz | 1.5 | 1.5 | 1.5 | |
| Reactor tube | ||||||
| Reactor length | H | cm | 265 | 150 | 150 | |
| Reactor tube diameter (external) | D outer | mm | 12 | 12 | 12 | |
| Reactor tube diameter (internal) | D internal | mm | 10 | 10 | 10 | |
| Reactor total volume | V tot | cm3 | 208 | 118 | 118 | |
| Reactor net volume | V net | cm3 | 131 | 78 | 78 | |
| Reactor contact area | A | cm2 | 622 | 471 | 471 | |
| MJOD unit | ||||||
| Total number of disks | N | 165 | 94 | 95 | ||
| Diameter of the disk | D disk | mm | 9.9 | 9.9 | 9.9 | |
| Thickness of the disk | h disk | mm | 4 | 4 | 4 | |
| Jet diameter | d jet | mm | 1.30 | 1.30 | 1.30 | |
| Number of jets of a disk | N jet | 4 | 4 | 4 | ||
| Distance between disks | h cavity | mm | 12 | 12 | 12 | |
| Piston shaft diameter | d | mm | 6 | 6 | 6 | |
| Reactor capacity/performance | ||||||
| Areal to volume ration | AV | m2 m−3 | 6.5 | 6.5 | 6.5 | |
| Flow reactor mass throughput | RMT | g min−1 | 0.26 | 1.46 | 2.26 | |
The reagents and substrate are prepared as solutions and transferred to dedicated reagent reservoirs. The flow reactor is preloaded with a suitable solvent. Each of the reservoirs (R) are connected to reagent pump (P) that supply the actual reagent at a pre-programmed rate.
The pump rates for each single pump are mutually adjusted to accomplish a chosen reactant ratio at the flow reactor input section. Moreover, the pump rates are likewise adjusted with respect to fulfill the desired reactor residence time. Due to the oscillating movement of the MJOD unit, “kick-back” to the feeding pumps can occur. In order to handle this problem, each input line (the tube from the pump to the reactor input section) were furnished with one-way valves. The post reaction mixture was collected at the output line at the output section of the reactor. The oscillator can be adjusted to deliver an amplitude A = 10–20 mm and an oscillator frequency f = 0–3 Hz.
Synthesis of 4,5-diiodo-1H-imidazole – batch protocol
A solution of imidazole (11.1 g, 0.163 mol) in NaOH (4.0 M, 600 mL) was added drop-wise to a solution of KI (147.2 g, 0.887 mol) and I2 (88.2 g, 0.348 mol) in water (500 mL) over a period of 30 min. The reaction mixture was stirred at room temperature for 24 h and then neutralized with acetic acid, which resulted in precipitation of the product. The mixture was cooled with an ice-water bath, filtered, and the product washed with several portions of ice-water and air dried to provide the title compound as creamy crystals in a yield of 82% (42.6 g, 0,134 mol); m.p. 185.7–186.6 °C. Rf = 0.47 [EtOAc/hexane (1:1)]. 1H NMR (400 MHz, d6-DMSO): δ (ppm) = 7.78 (s, 1 H). 13C NMR (d6-DMSO): δ (ppm) = 143.0, 141.6.
Synthesis of 4(5)-iodo-1H-imidazole – batch protocol
A solution of 4,5-diiodo-1H-imidazole (38.0 g, 0.119 mmol) and K2SO3 (186 g, 1.17 mol) in ethanol (30% in water, 400 mL) was stirred and heated at reflux for 24 h. The reaction mixture was then cooled at room temperature and the inorganic salts were filtered off. Ethanol was removed under reduced pressure whereupon the water phase was saturated with sodium chloride. The water phase was then extracted with ether/tetrahydrofuran (1:1; 3 × 300 mL). The organic layers were combined and washed with small portions of K2SO3 solution (sat.) until the yellow color disappeared. The organic extract was dried over anhydrous sodium sulfate, filtered, and the solvent removed under reduced pressure to provide the title compound as a white solid in a yield of 96% (22.2 g, 0.114 mol); m.p. 137.9–138.3 °C. Rf = 0.47 [EtOAc/hexane (1:1)]. 1H NMR (400 MHz, CDCl3): δ (ppm) = 7.17 (s, 1 H), 7.62 (s, 1 H).
Flow synthesis of 4,5-diiodo-1H-imidazole
The flow reactor MJOD1 [section (a) of Fig. 2] was prepared by pre-cooling the reactor body at 0 °C. The MJOD oscillator (O1) was adjusted at an oscillating rate of f = 1 Hz. Imidazole (0.655 g, 9.62 mmol) was dissolved in a mixture of H2O (20 mL) and H2SO4 (10 mL) and transferred to reservoir R1. N,N-diiodo-5,5-dimethylhydantonin (5.621 g, 14.8 mmol) was dissolved in methanol (8 mL) and then transferred to reservoir R2. The two reservoirs were connected to pumps P1 and P2, respectively. The reagent and substrate mixtures of the two reservoirs were pumped into the MJOD reactor input section at rates of 1.27 mL min−1 and 0.35 mL min−1 providing a residence time of 15 min. The reaction mixture was quenched in-line by pumping (P3) ice-cold aqueous sodium hydroxide (7 M, 50 mL) from a reservoir (R3) (2.17 mL min−1). The quenched reaction mixture was collected in a hold-up tank (R5a,b-STR) and neutralized with glacial acetic acid (R4 connected to P4) (pH ≈ 6–7) whereupon 4,5-diiodo-1H-imidazole precipitated. The slurry was then pumped (P5) to a filter unit (F1a,b) and the solid target product was isolated by means of a Büchner funnel furnished with filter paper. The isolated product was then washed with ice-cold water (3 × 5 mL) and air dried. Target 4,5-diiodo-1H-imidazole was isolated in a quantitative yield (9.60 mmol, 3.060 g). 1H NMR (400 MHz, d6-DMSO): δ (ppm) = 12.9 (1H, br, s), 7.7 (1H, s). FTIR (neat): ν = 3112 (w), 3076 (w), 2956 (w), 2774 (m), 2580 (m), 1807 (m), 1641 (m), 1538 (m), 1452 (m), 1283 (m), 1177 (m), 1149 (m), 953 (s), 915 (s), 815 (s). MS (EI) m/z (%) = 319.9 (100), 253.8 (6), 193 (82), 165.9 (53), 126.9 (33), 67.1 (2), 66.0 (8).Flow synthesis of 4(5)-iodo-1H-imidazole HCl – protocol for the statistical experimental design
The flow reactor MJOD1 was prepared by pre-cooling the reactor body at 0 °C and the oscillator (O1) was adjusted and controlled to provide an oscillating rate of f = 0.5 Hz. Imidazole (1.498 g, 22 mmol) was dissolved in a mixture of H2O (17 mL) and H2SO4 (3 mL) and added to reservoir (R1). N,N-diiodo-5,5-dimethylhydantonin (3.142 g, 8.27 mmol) was dissolved in methanol (18 mL) and added to reservoir R2. Reservoirs R1 and R2 were connected to their corresponding pumps P1 and P2. The reagents were pumped into the MJOD reactor at 1.14 mL min−1 and 1.03 mL min−1 providing a reactor residence time of 30 min. The reactor outlet was directed to a quench solution composed by an ice-cold NaOH solution (7 M, 50 mL). The quenched reaction mixture was subsequently neutralized with glacial acetic acid (pH ≈ 6–7) and saturated with NaCl. This treated post reaction mixture was then extracted with diethyl ether (5 × 40 mL) whereupon the organics was combined and extracted using HCl solution (10%, 5 × 10 mL). The aqueous HCl was then removed under reduced pressure to provide target 4(5)-iodo-imidazol hydrochloride in a yield of 65.3% (2.462 g, 10.8 mmol). 1H-NMR (500 MHz, D2O): δ (ppm) = 8.6 (1H, s), 7.4 (1H, s). 13C-NMR (125 MHz, D2O): δ (ppm) = 136.6, 125.7 67.6. FTIR (neat): ν = 3216 (w), 3122 (w), 2945 (w), 2873 (w), 2757 (m), 2570 (m), 2163 (m), 1749 (w), 1622 (w), 1572 (m), 1431 (m), 1260 (w), 1146 (m), 1073 (m), 933 (s), 827 (s), 789 (s), 692 (s).Flow synthesis of 4(5)-iodo-1H-imidazole hydrochloride concatenated with continuous work-up steps
The flow reactor MJOD1 [section (a) of Fig. 7] was cooled at 0 °C and the oscillator (O1) was adjusted at a frequency of f = 1.5 Hz. Imidazole (6.00 g, 88 mmol) was dissolved in a mixture of H2O (68 mL) and H2SO4 (11 mL) and transferred to the reservoir R1. N,N-diiodo-5,5-dimethylhydantonin (10.1 g, 26.6 mmol) was dissolved in methanol (72 mL) and transferred to reservoir R2. The contents of reservoirs R1 and R2 was pumped (P1 and P2) into the flow reactor MJOD1 at rates 1.33 mL min−1 and 1.20 mL min−1, respectively. The feeding rates were adjusted in order to achieve a residence time of 30 min. Aqueous NaOH (7 M) was transferred to reservoir R3 and pumped (P3) into the upper section of the flow reactor MJOD1 at a rate of 2.67 mL min−1 that provided a reaction quenching time of 3.6 min. The flow reactor (MJOD1) outlet was directed to the hold-up tank (5a,b-STR) of V = 2000 mL (several stirred tank reactors was coupled in parallel allowing a continuous production in the flow reactor). Glacial acetic acid from reservoir R4 was pumped (P4) to neutralize (pH ≈ 6–7) the mixture, whereupon the mixture was saturation by adding NaCl (S1). The MJOD2 reactor was prepared for liquid–liquid extraction using an oscillation frequency of f = 1.5 Hz (O2). Diethyl ether was added to reservoir R6 and pumped (P6) into the flow reactor (operating as an extractor) MJOD2 together with the contents of the hold-up tank at rates 7.67 mL min−1 and 3.83 mL min−1, respectively. The flow rate of the flow reaction (MJOD2) corresponds to a residence time of 5.4 min. The reactor outlet was directed to a liquid–liquid separator (R7-LS1, V = 1000 mL) where the aqueous phase was removed. The flow reactor MJOD3 operated as a continuous flow liquid–liquid extractor using an oscillation frequency (O3) of f = 1.0 Hz. A HCl solution (10%) was transferred to reservoir R8 and pumped (P8) into the MJOD3 flow reactor together with the contents of R7 (organic phase) at the rates 5.88 mL min−1 and 11.75 mL min−1, respectively, which provided a residence time of 3.5 min. The reactor output (from MJOD3) was directed to a liquid–liquid separator (R9-LS2) where the organic phase was removed for solvent recovery (containing 5,5-dimethylhydantoin as waste) and the aqueous phase was transferred by a pump (P9) or drawn into the rotary evaporator that was placed under reduced pressure in order to remove the HCl solution.An ultimate experimental run using the above settings was performed for a production period of 120 min. This run provided 7.9 g (34.65 mmol) of title compound, a quantity that corresponds to an isolated yield of 65%. The concatenated overall process produced target compound at a rate of ≈0.066 g min−1 (3.95 g h−1).
Reactor specification
The MJOD flow reactor that was used for the continuous flow iodination was assembled with two sections; the bottom (lower) section constitutes a length of HL = 196 cm that correspond to a volume of VL = 76 mL. The upper section was reactor tube with a length of HU = 68 cm and VU = 26 mL. The entire flow reactor was cooled at T = 0 °C. The MJOD oscillator was adjusted to a frequency f = 1 Hz with an oscillating amplitude of A = 20 mm.The MJOD reactor used for the continuous flow liquid–liquid extraction was composed of one length Hextract = 150 cm, which correspond to a net volume of Vextract = 62 mL. The MJOD oscillator was adjusted to a frequency f = 1.5 Hz with an amplitude A = 20 mm.
Reservoir R1: Imidazole (1.498 g, 22 mmol) was dissolved in a mixture of H2O (17 mL) and concentrated sulfuric acid (3 mL). Reservoir R2: DIH (3.340 g, 8.8 mmol) was dissolved in methanol (18 mL). The two reservoirs R1 and R2 were connected to pump P1 and P2, respectively.
The reagents were pumped at rate of r1 = 1.33 mL min−1 and r2 = 1.20 mL min−1. At the inlet of the upper section (30 min residence time) the reaction mixture was quenched inline by a 3 M NaOH solution (40 mL, 2.67 mL min−1). The quenched reaction mixture was then collected in a holding tank where it was neutralized with acetic acid and saturated with NaCl. The holding tanks content was pumped (3.87 mL min−1) into a MJOD extractor together with diethyl ether (7.67 mL) that gave an extraction time of 5.37 min. The water was then removed with a separation unit. The organic phase was then subjected to an acid workup with 10% HCl solution (4 × 20 mL), aquatic phase was then collected and solvents removed, giving the desired product.
Investigation of the extraction efficiency
Samples of the post-reaction mixture (4 × 100 mL) were withdrawn from the hold-up tank (R5a/b). These samples were subjected to various work-up trials in order to benchmark the extraction efficiency of the flow reactor (MJOD2) as an continuous liquid–liquid flow extractor versus the classical repeated laboratory batch extractions using a separatory funnel (entry 4, Table 4). The experimental set-up with adjacent experimental results is summarized in Table 4. The extraction efficiency relative to the classical batch extraction method is expressed as an extraction efficiency, η-%, in the bar graph of Fig. 8. At a ratio 1:2 of post reaction mixture (PRM) to extraction solvent at flow rates 3.83 and 7.67 mL min−1, respectively.
| # | Ratioa PRM:ES |
Flow rateb [mL min−1] | Y [g] | |
|---|---|---|---|---|
| PRM | ES | |||
| a PRM = post reaction mixture (from the continuous reaction flow step) and ES = extraction solvent. b BEM = batch extraction method and ES = solvent. c y = isolated yield [g]. | ||||
| 1 | 1: 1 | 5.75 | 5.75 | 0.593 |
| 2 | 1: 1.5 | 4.60 | 6.90 | 0.638 |
| 3 | 1: 2 | 3.83 | 7.67 | 0.898 |
| 4 | 1: 2 | BEM | 0.795 | |
 | ||
| Fig. 8 The graph shows the extraction efficiency using various volume ratios of the post reaction mixture (PRM) versus the extraction solvent (ES) at a fixed flow rate of 11.2 mL min−1 in each continuous flow extraction experiment. At a ratio PRM:ES = 1:2, a more efficient continuous extraction process is achieved compared with the batch extraction protocol [PRM:ES = 100 mL:40 mL × 5 ]. | ||
This MJOD continuous flow liquid–liquid extraction [Fig. 7(b)] was successfully introduced as a work-up step in the process. By means of an identical solvent volume (as in batch), the continuous flow extraction process afforded an improved efficiency of 13 percentage points (Fig. 8). Furthermore, the extraction method requested a residence time of 5.6 min, which result in a substantial decreased overall processing time.
Conclusions
We have developed and optimized multi-gram continuous flow processes for the preparation of 4,5-diiodo-1H-imidazole 2 and 4(5)-iodo-1H-imidazole 3 and 3·HCl. During the development phase we isolated multi-grams quantities of high quality products of our target products, namely >95 g of 3·HCl and ≈125 g of 2. The established continuous flow process for the preparation of product 3 involved several work-up steps and revealed excellent properties of the MJOD flow reactor as a liquid–liquid extractor. The process leading to product 2 included a semi-continuous work-up step, where product 2 precipitated/crystallized during the course of the process and isolated as a pure product using a semi-continuous vacuum filtration step installed at the outlet of the MJOD flow reactor.Acknowledgements
A. D. is grateful to the Department of Chemistry, University of Bergen for his research fellowship. A. D. acknowledges Dr. Alexander H. Sandtorv for useful and detailed advices regarding the previously published batch protocols.Notes and references
- (a) M. S. R. Naidu and H. B. Bensusan, J. Org. Chem., 1968, 33, 1307 CrossRef; (b) S. J. Garden, J. C. Torres, S. C. de Souza Melo, A. S. Lima, A. C. Pinto and E. L. S. Lima, Tetrahedron Lett., 2001, 42, 2089–2092 CrossRef CAS; (c) N. Matsunaga, T. Kaku, A. Ojida and A. Tasaka, Tetrahedron: Asymmetry, 2004, 15, 2021–2028 CrossRef CAS; (d) A. Kirschning, M. S. Yusubov, R. Y. Yusubova, K.-W. Chi and J. Y. Park, Beilstein J. Org. Chem., 2007, 3(19) DOI:10.1186/1860-5397; (e) S. V. Bhilare, A. R. Deorukhkar, N. B. Darvatkar and M. M. Salunkhe, Synth. Commun., 2008, 38, 2881–2888 CrossRef CAS; (f) A. H. Sandtorv and H.-R. Bjørsvik, Adv. Synth. Catal., 2013, 355, 499–507 CrossRef CAS.
- (a) A. H. Sandtorv and H.-R. Bjørsvik, Adv. Synth. Catal., 2013, 355, 3231–3243 CrossRef CAS; (b) A. H. Sandtorv, K. W. Törnroos and H.-R. Bjørsvik, Eur. J. Org. Chem., 2015, 3506–3512 CrossRef CAS; (c) A. H. Sandtorv and H.-R. Bjørsvik, Eur. J. Org. Chem., 2015, 4658–4666 CrossRef CAS.
- A. H. Sandtorv, C. Leitch, S. L. Bedringaas, B. T. Gjertsen and H.-R. Bjørsvik, ChemMedChem, 2015, 10, 1522–1527 CrossRef CAS PubMed.
- (a) Mictroreactors in Organic synthesis and catalysis, ed. T. Wirth, Wiley-VCH, New York, Weinheim, 2009 Search PubMed; (b) S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed; (c) S. V. Ley, D. E. Fitzpatrick, R. M. Myers, C. Battilocchio and R. J. Ingham, Angew. Chem., Int. Ed., 2015, 54, 10122–10136 CrossRef CAS PubMed.
- M. Ferreri, A. Drageset, C. Gambarotti and H.-R. Bjørsvik, Continuous flow synthesis telescoped with semi-continuous product isolation: Multi-100 g preparation of 1,3-diiodo-5,5-dimethylimidazolidine-2,4-dione, React. Chem. Eng., 2016 10.1039/c6re00051g.
- L. Liguori and H.-R. Bjørsvik, Org. Process Res. Dev., 2011, 15, 997–1009 CrossRef CAS.
- D. L. Browne, B. J. Deadman, R. Ashe, I. R. Baxendale and S. V. Ley, Org. Process Res. Dev., 2011, 15, 693–697 CrossRef CAS.
- R. L. Hartman, Org. Process Res. Dev., 2012, 16, 870–887 CrossRef CAS.
- Fluid Metering Inc., USA, supplied the slurry-feeding pump. The pump was a valve less ceramic piston pump (Q1CKCW) that is suitable for pumping saline, abrasives, particulates, anaerobic, and crystal forming fluids. The pump head is furnished with two extra ports for gland “barrier” liquid of gas.
- T. Horie, M. Sumino, T. Tanaka, Y. Matsushita, T. Ichimura and J. Yoshida, Org. Process Res. Dev., 2010, 14, 405–410 CrossRef CAS.
- G. E. P. Box, W. G. Hunter and J. S. Hunter, Statistics for experimenters. Design, Innovation, and Discovery, 2nd edn, Wiley, New York, 2005 Search PubMed; G. E. P. Box and N. R. Draper, Empirical model-building and response surfaces, Wiley, New York, 1987 Search PubMed; G. E. P. Box, Improving almost anything. Ideas and essays, rev. edn, Wiley, New York, 2006 Search PubMed.
- N. R. Draper and H. Smith, Applied Regression Analysis, 3rd edn, Wiley, New York, 1998 Search PubMed; D. C. Montgomery and E. A. Peck, Introduction to linear regression analysis, Wiley, New York, 1982 Search PubMed.
- K. Ishikawa, Guide to quality control, Chap 3. Cause-and-effect diagram, Asian Productivity Organization, 1982, pp. 18–29 Search PubMed.
- Fractional factorial design (ffd) 24–1 of resolution R=IV implies that the main effects are aliased with the three-variable (factor) interactions, which however is considered to negligible. The two-variable interactions are aliased with one another according to the following pattern: β12+β34, β13+β24, and β14+β23. In order to create the ffd: 2,4–1 a generator x4 = x1 × x2 × x3 was used in addition to a standard order design for the variables x1, x2, x3. See ref. 11a pp. 240–244 and p. 272.
- The MATLAB program was utilized for the graphical representation of the results and models, see (a) Using MATLAB Version 6 (Nov. 2000), The MathWorks, Inc., Natick, MA, USA Search PubMed; (b) Using MATLAB Graphics Version 6 (Nov. 2000), The MathWorks, Inc., Natick, MA, USA Search PubMed; (c) A. Briran and M. Breiner, mMATLAB for Engineers, Addison-Wesley, Wokingham, England, 1995 Search PubMed; (d) D. Hanselman and B. Littlefield, Mastering MATLAB. A Comprehensive Tutorial and Reference, Prentice-Hall, Upper Saddle River, NJ, USA, 1996 Search PubMed.
- (a) C. Daniel, Technometrics, 1959, 1, 311–341 CrossRef; (b) C. Daniel, Application of Statistics to Industrials Experimentation, Wiley, New York, 1976 Search PubMed; (c) See also ref. 11a, pp. 33–34 and pp. 203–205.
- (a) C. H. Hornung, M. R. Mackley, I. R. Baxendale and S. V. Ley, Org. Process Res. Dev., 2007, 11, 399–405 CrossRef CAS; (b) L. Di Marco, M. Hans, L. Delaude and J.-C. M. Monbaliu, Chem. – Eur. J., 2016, 22, 4508–4514 CrossRef CAS PubMed; (c) M. O′Brien, P. Koos, D. L. Browne and S. V. Ley, Org. Biomol. Chem., 2012, 10, 7031–7036 RSC; (d) D. X. Hu, M. O′Brien and S. V. Ley, Org. Lett., 2012, 14, 4246–4249 CrossRef CAS PubMed; (e) L. Osorio-Planes, C. Rodrígez-Escich and M. A. Pericàs, Catal. Sci. Technol., 2016, 6, 4686–4689 RSC; (f) A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J. –C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, Science, 2016, 354, 61–65 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6re00091f |
| This journal is © The Royal Society of Chemistry 2016 |