
A reactive continuous chromatographic process for protein PEGylation
Oliver
Ingold
,
David
Pfister
and
Massimo
Morbidelli
*
Institute of Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zurich, 8093 Zurich, Switzerland. E-mail: massimo.morbidelli@chem.ethz.ch
First published on 4th January 2016
Abstract
In a previous work, a batch on-column PEGylation process was developed and thoroughly investigated (Pfister et al., Reaction Chemistry & Engineering, 2016). A mathematical model was derived and its predictions compared with experimental data were obtained under various conditions. As an alternative, a multicolumn counter-current chromatographic setup is considered to turn the batch on-column PEGylation process into a continuous one. Using the parameters previously estimated, the continuous chromatographic process in the form of a reactive multicolumn counter-current solvent gradient purification (rMCSGP) process was simulated and the performance of the process was assessed. Batch experiments were performed to show the feasibility of the method and to determine the parameters specific to the design of multicolumn processes. It is shown that the rMCSGP process can improve the throughput, purity and yield for the production of mono-PEGylated protein.
Introduction
PEGylation is a hot topic for the pharmaceutical industry due to its ability to improve the in vivo circulation half-life of proteins.1 To date, more than a dozen of PEGylated drugs are on the market and more are entering clinical trials.2 Among the marketed drugs, half of them are still produced by non-selective lysine PEGylation and till now, PEGylation of amino groups remains the most frequently used PEGylation technique. On the one hand, this strategy is versatile thanks to the natural abundance of lysine residues. On the other hand, the large abundance of this amino acid causes selectivity issues. PEGylation at the lysine residues generates mixtures of conjugates with different degrees of PEGylation, each one composed of multiple positional isomers.3To improve the selectivity of the reaction, PEGylation can be performed in a chromatographic column. The on-column PEGylation strategy can potentially avoid the formation of multi-PEGylated species by reducing the number of accessible reactive residues and therefore can lead to more selective formation of specific positional isomers. Indeed, once adsorbed, some of the PEGylation sites facing the adsorbent surface become inaccessible, thus reducing the probability to obtain multi-PEGylated products. Moreover, the integration of the reaction in the chromatographic column reduces the number of steps so that PEGylation and separation can be achieved within the same unit. Integrated processes combining reaction and separation are promising options to save time and equipment. This strategy has already been investigated by several authors who all reported some improvements compared to the classical solution PEGylation reaction.4–6
In a previous work, we reported the on-column PEGylation of lysine groups of α-lactalbumin adsorbed on a Sepharose strong anion-exchange resin.7 The integration of the reaction in the chromatographic column makes the process easier to operate, faster and economically interesting. This process was thoroughly investigated and a more fundamental understanding of the kinetics and the influence of process parameters was obtained by means of a mathematical model. A reactive chromatographic model was derived and by comparison with experimental data, important model parameters (adsorption parameters and on-column kinetic rate constants) were estimated.
Switching from the batch process to the continuous production of biopharmaceuticals can potentially increase the productivity and the robustness of processes while reducing the footprint and the capital investment.8 Herein, the implementation of the previously developed on-column process using a reactive multicolumn counter-current solvent gradient purification (rMCSGP) process is reported. A three-column setup performing simultaneously the on-column PEGylation reaction and the product purification is described. Thanks to the internal recycling of the non-resolved fractions and unreacted protein, the yield of the mono-PEGylated protein can be increased together with the conversion. A high yield of the mono-PEGylated protein together with a high conversion of the protein can be obtained simultaneously with this rMCSGP process.
Experimental section
Materials
α-Lactalbumin, 85% pure, was purchased from Sigma-Aldrich and mPEG-SPA (10 kDa) was purchased from JenKem Technology. All the buffer components (reagent grade) were purchased from Sigma-Aldrich. The chromatographic resin Q Sepharose HP was purchased from GE Healthcare and packed in a Tricorn column according to the manufacturer's protocol. The final bed dimensions are 143 × 5 mm.Batch on-column PEGylation
The kinetic study performed using the batch on-column PEGylation process can be found in our previous work.7 The PEGylation protocol is identical and is briefly discussed in this section.The resin of choice for this reaction is the Q Sepharose HP resin. The 143 × 5 mm column was pre-equilibrated with 25 mM Tris-HCl buffer solution adjusted to pH 7 and maintained at 25 °C. 1 mg of protein, solubilized in 2.5 mL of 25 mM Tris-HCl, was injected into the column by means of a manual injection valve fitted with a 2.5 mL external loop (Rheodyne). The unbound material was eluted with 5 mL of the same buffer solution at 1 mL min−1. 2.5 mL of mPEG-SPA solution containing a 16-fold molar excess of mPEG-SPA with respect to the loaded protein was injected into the external loop and then slowly fed into the column at a flow rate of 0.03 mL min−1. To slow down the functionalized PEG hydrolysis reaction, the external loop was maintained at 0 °C in an ice-cold water bath. After complete injection of the PEG solution, the column was washed with the 25 mM Tris-HCl buffer at pH 7.0 for 10 minutes at 1 mL min−1 to get rid of the NHS by-product and the unreacted and hydrolyzed PEG. Then, the modifier (i.e. NaCl) concentration was suddenly increased to 50 mM and maintained at this level for 10 minutes. After this, the linear salt gradient started and the modifier concentration was increased to 176 mM over 9 minutes. The concentration of the modifier was finally reduced to 0 mM and the resin was re-equilibrated with the starting buffer.
Characterization
Size exclusion chromatography analyses were performed using a Tricorn Superdex 200 10/300 GL high performance column (GE Healthcare). The mobile phase was a 25 mM Na2HPO4/100 mM Na2SO4 solution adjusted to pH 7.0. After calibration with standard proteins, the hydrodynamic radii of proteins and PEGylated proteins were estimated.For the mass spectrometry analysis, the samples were desalted and concentrated by using a ZipTip C4. Matrix-assisted laser desorption/ionization time of flight (MALDI-ToF) mass spectrometry was performed on a Bruker Daltonics Ultraflex mass spectrometer. Matrix: 2,5-DHAP (7.6 mg ml−1) in 375 μl of EtOH mixed with 125 μl of DAC (18 mg ml−1) in H2O. 1 μl of the sample solution was mixed with 1 μl of 2% TFA and 1 μl of the matrix solution. 2 μl was spotted on target and analyzed by MALDI-MS in the linear mode.
Process description
Cycles, switches and phases
The continuous chromatographic process used in this work was in the form of a multi-column counter-current solvent gradient purification (MCSGP) unit. This continuous multi-column chromatographic process9 can perform three-fraction separation while using a linear modifier concentration gradient to achieve this separation. It is particularly convenient for the separation and fractionation of three-solute mixtures in a single unit. This makes this process particularly well suited for the purification of mono-PEGylated protein from a mixture containing the unreacted protein together with multi-PEGylated products. To save stationary phase material and equipment, the original MCSGP process involving up to six columns10,11 was simplified and recently a twin-column setup was developed.12 This simplification is necessarily obtained at the expense of the process continuity. The semi-continuous twin-column MCSGP process is a hybrid multi-column process oscillating between two different states:13–15 a first one in which the solutes are collected and a second one where the non-resolved fractions are internally short-circuited and recycled in one of the two columns. The two columns therefore alternate between a disconnected elution phase and an interconnected recycling phase during which the product of interest co-elutes with impurities. The twin-column MCSGP is described in detail in the works of Krättli et al.12 and Müller-Späth et al.14,15For the reactive MCSGP process (rMCSGP), a 3-column setup was implemented. Technically speaking, the process is a twin-column MCSGP process (only 2 columns) fitted with an additional column dedicated to the on-column PEGylation reaction. In this (2 + 1) multi-column process, the different columns switch from the ‘reaction’ position to the ‘capture’ position, and finally to the ‘elution’ position. A schematic flowchart of the process, representing the three columns during a switch time, is presented in Fig. 1.
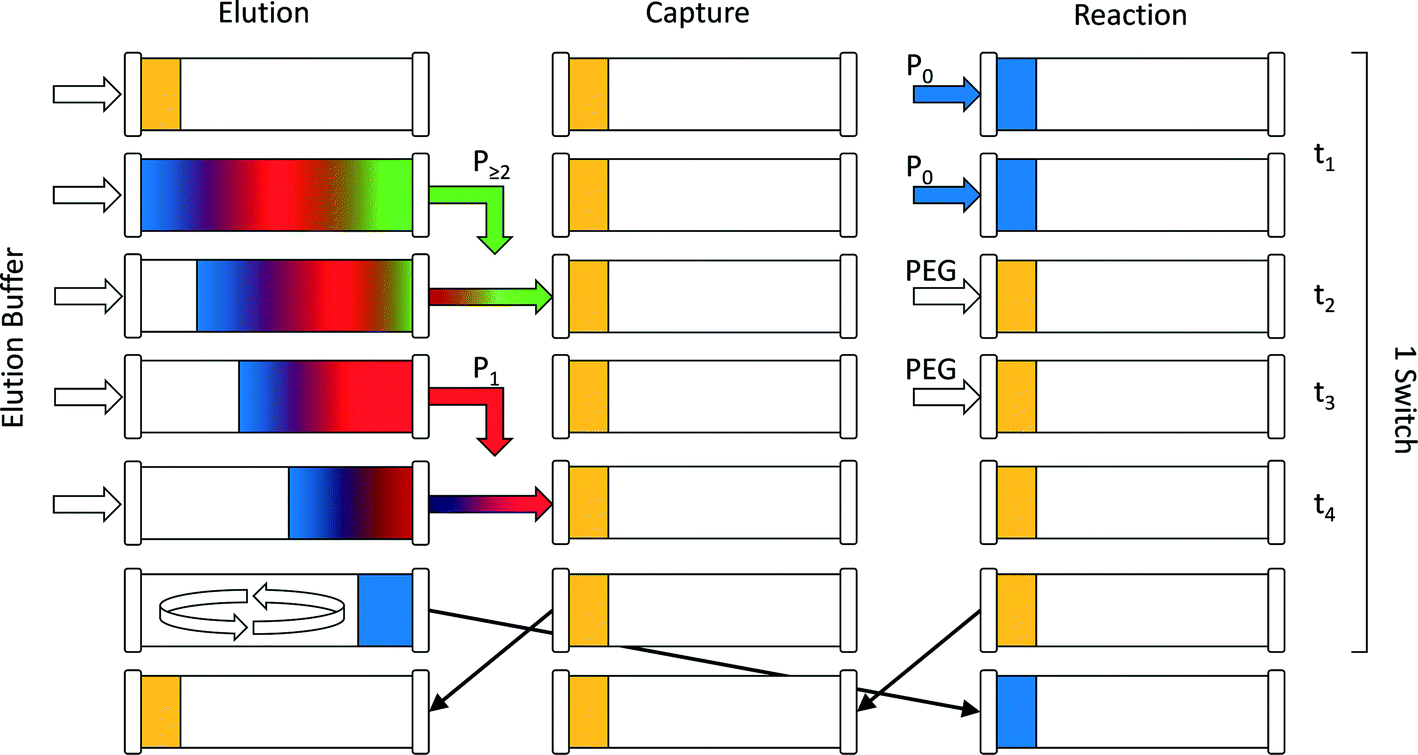 | ||
| Fig. 1 Schematic representation of one switch time in the (2 + 1)-column rMCSGP setup. | ||
The process time is divided into cycle times, i.e. the time needed before one column goes back to its initial role, and switch times, i.e. the time spent at one position. Moreover, each switch time is divided into four phases whose times are denoted as ti, where i = 1–4. According to these definitions, one cycle is composed of three switches and each one of them is divided into 4 phases. During one switch time, the columns have the task of either PEGylating the adsorbed protein (‘reaction’ position), capturing the non-resolved overlapping fractions (‘capture’ position) or separating the product mixture (‘elution’ position). The three tasks are presented in Fig. 1 and correspond to the three columns while the different phases are depicted in rows. At the end of each switch time (i.e. at the end of phase 4), the columns virtually rotate in their position, which is made possible by proper valve position switching, in the order: reaction/capture/elution and back to the reaction position. The design of the different phases is crucial for this process and is obtained according to the method developed by Müller-Späth et al.16 This method will be further explained in the next section of this work.
To better understand the rotation of the column positions, let us follow one column through three switches. The complete cycle is shown in Fig. 2. In the initial configuration (i.e. during the first switch time), the three columns denoted as 1, 2 and 3 in Fig. 2 correspond to the positions ‘reaction’, ‘capture’ and ‘elution’ in Fig. 1, respectively. At the beginning of the sequence, the protein is fed into the ‘reaction’ column and adsorbs onto the resin (see column 1 in the first switch time). A solution of mPEG-SPA is then injected into this column and the on-column PEGylation reaction takes place. After this, the ‘reaction’ column (1) is washed as described in the experimental section, while the protein and the products of the reaction remain adsorbed. In the next step of the sequence, by means of proper valve position switching, the ‘reaction’ column (1) takes the position of the ‘capture’ column (2) which, in turn, becomes the ‘elution’ column (3).
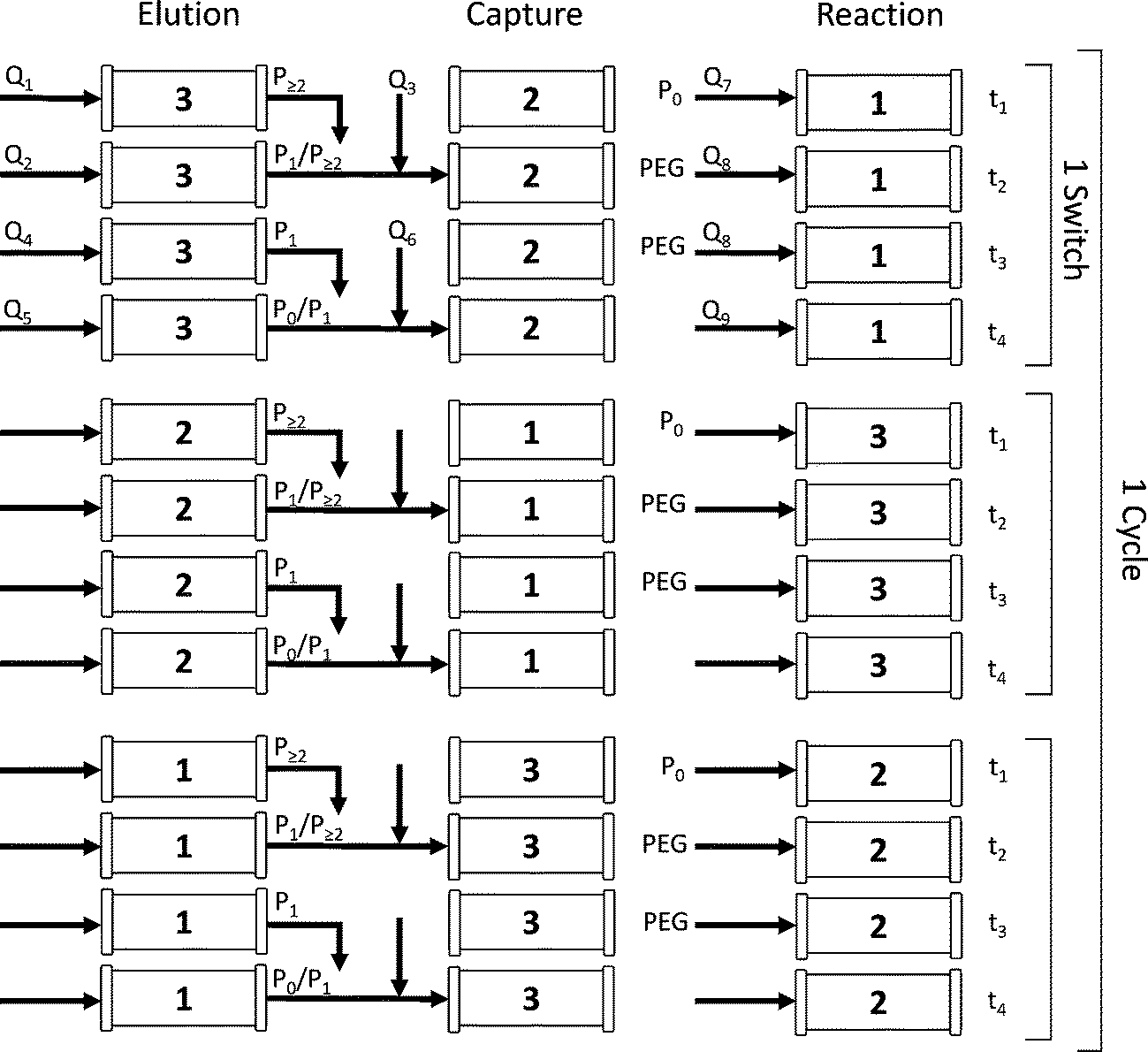 | ||
| Fig. 2 Flow diagram of the reactive MCSGP process during one cycle time. Pi corresponds to the protein with i conjugated PEG chains and Qi is a volumetric flow rate. | ||
The different phases are defined with respect to this ‘elution’ column in which the solutes elute under the effect of the modifier concentration gradient and are collected. In Fig. 2, for the first switch time, it can be seen that phase 1 corresponds to the elution of the weakly adsorbing solute, which in the present case is essentially composed of di-PEGylated protein. Phase 2 is the first interconnected phase: the stream eluting from the ‘elution’ column (3) contains some multi-PEGylated proteins co-eluted with the product of interest (i.e. the mono-PEGylated protein). This fraction is recycled into the ‘capture’ column (2). Phase 3 corresponds to the elution of the product. And finally, phase 4 is equivalent to phase 2, except that the recycled stream contains the product co-eluted with the unreacted protein. In the end, the ‘capture’ column (2), which has now a large amount of product and impurities adsorbed in it (i.e. the product of the PEGylation reaction in addition to the solutes internally recycled), enters the ‘elution’ position (3).
At the end of phase 4, the modifier gradient is prematurely stopped in the ‘elution’ column (3), before the unreacted protein starts eluting. The flow direction in this column is then reversed before the column goes back into the ‘reaction’ position. By doing so, the protein stays in the column, ensuring the internal recycling of the unreacted solute.
When this sequence ends, the process is ready to enter the second switch time. The former ‘elution’ column (see column 3) is now in the ‘reaction’ position and contains the internally recycled unreacted protein. Only the make-up of fresh protein is loaded into this column (3) to maintain a constant protein concentration in the system. The schematic representation of a full cycle (composed of 3 switch times) is shown in Fig. 2.
Model development
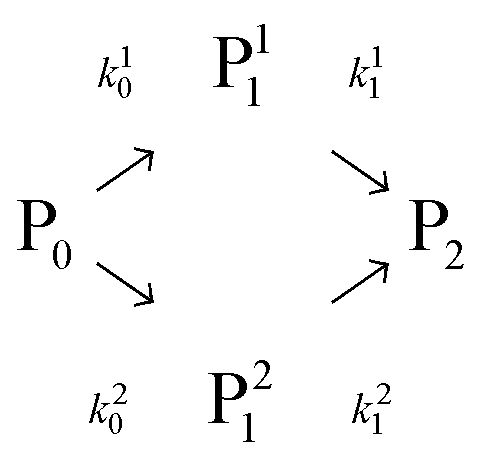 | (1) |
In this scheme, P0 is the α-lactalbumin protein. In the following, it will be assumed that k10 = k20 and k0 = k10 + k20. Therefore, the two mono-PEGylated PEGamers P11 and P21 are assumed to be formed at the same rate but differ in their reactivity (k11 and k21) to form the di-PEGylated species, P2.
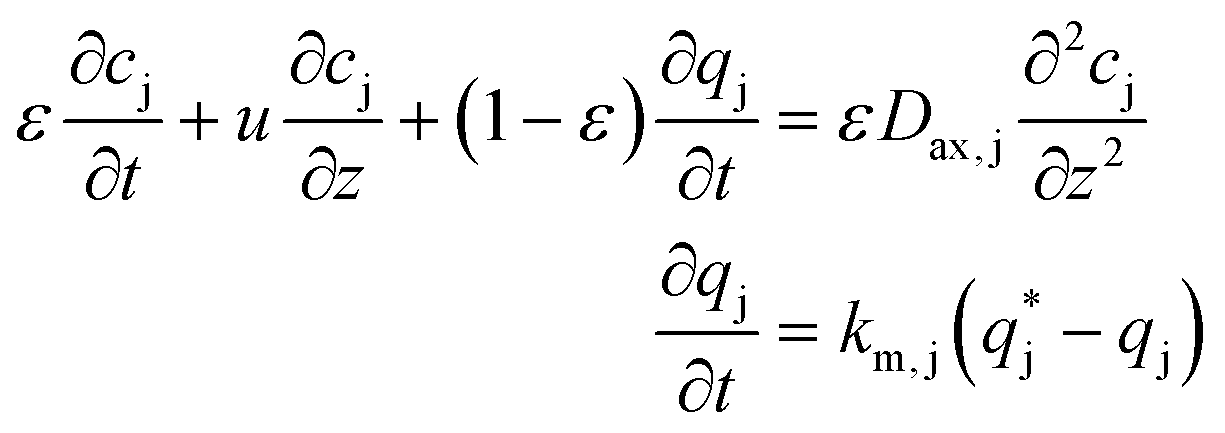 | (2) |
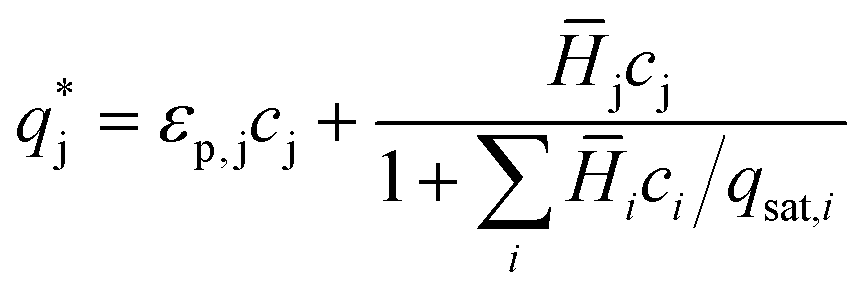 | (3) |
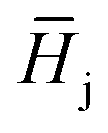 , is described in the frame of the stoichiometric displacement model:20
, is described in the frame of the stoichiometric displacement model:20 | (4) |
 ). The boundary conditions of eqn (2) are the following:
). The boundary conditions of eqn (2) are the following:
at
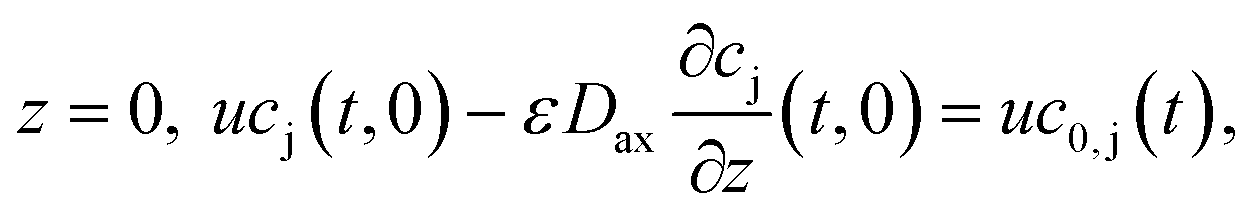
where c0,j (t) is the feed concentration of the solute j. Therefore, the initial conditions are defined as follows:
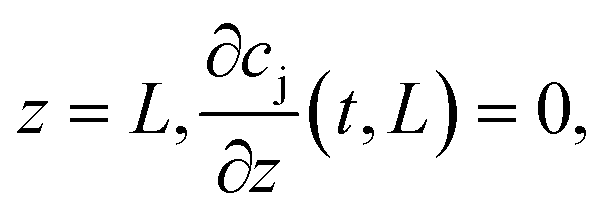
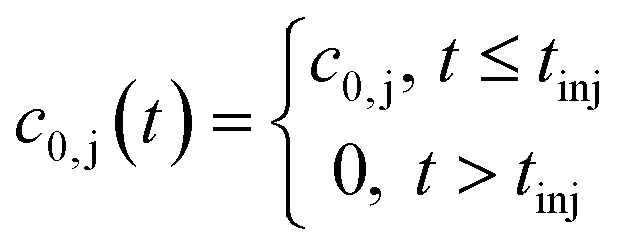 | (5) |
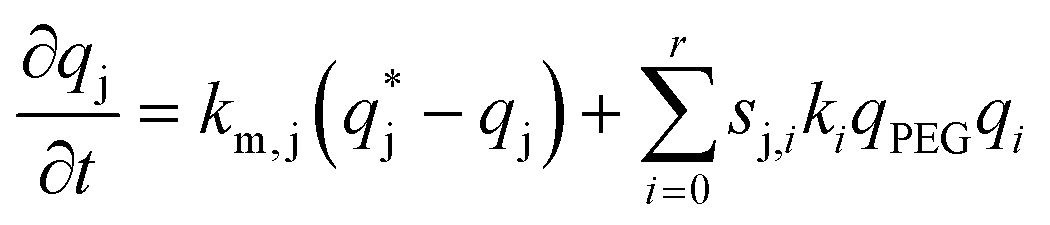 | (6) |
For the mPEG-SPA mass balance equation, it should be noted that the functionalized PEG is not only consumed by all the PEGylation reactions but also undergoes hydrolysis of its active moiety, so its mass balance equation has to be modified as follows:
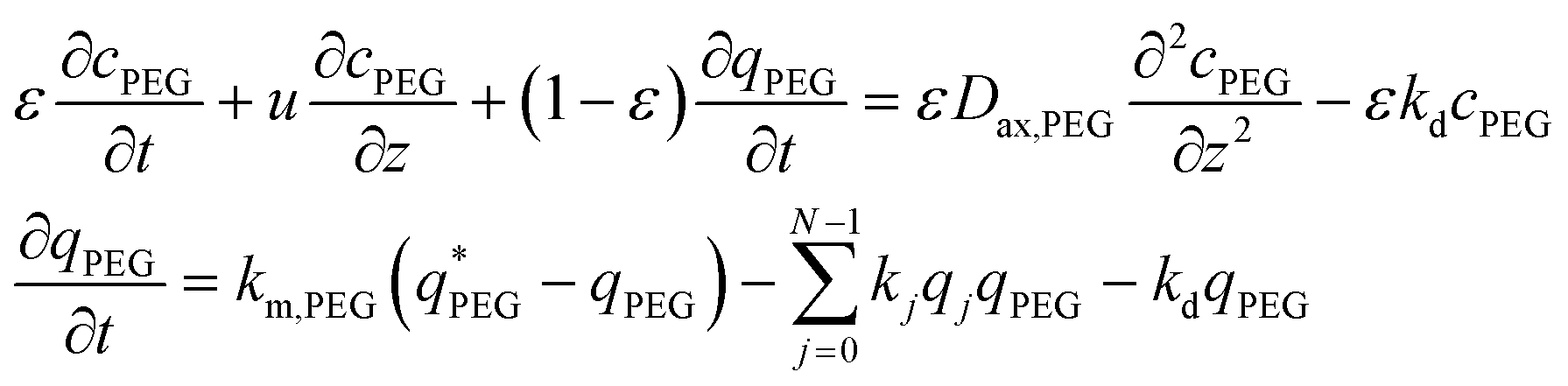 | (7) |
The chromatographic model was discretized using the finite difference method along the column length. The resulting set of ordinary differential equations was integrated with time using the subroutine DLSODE from the ODEPACK coded in FORTRAN.
Results and discussion
Batch on-column PEGylation
In order to validate the on-column PEGylation model, a reference batch on-column PEGylation reaction was performed by loading 1 mg of protein into the chromatographic column and injecting a 16-fold molar excess of mPEG-SPA solution over 80 minutes. At the end of the reaction, the products were eluted using the protocol previously described in the ‘Experimental’ section. The experiment was repeated twice and the mean values and standard deviations were computed from the two repetitions. This experiment was simulated with the on-column PEGylation model described above and the parameters estimated in our previous work.7The model parameters are recalled in Table 1. The comparison between experimental data and the result of the simulations is shown in Fig. 3. It can be seen that the model predictions are in good agreement with the experimental data, thus confirming the validity of the model and the associated model parameters. As expected, degrees of PEGylation up to two are observed. Moreover, two distinct peaks corresponding to the mono-PEGylated α-lactalbumin are identified. These two peaks correspond to two different subgroups of positional isomers identified as P11 and P21, P11 being the peak eluting the latest between the two. Due to the difficulty in separating these two subgroups of positional isomers and quantifying them separately, small discrepancies between experimental and simulated data can be seen in Fig. 3.
| Chromatography | K ∞ | log10 (a) | β | q sat | k m | D ax | |
| — | — | — | mg ml−1 | min−1 | cm2 min−1 | ||
| P0 | 1.47 | 8.6 | 3.7 | 218 | 10.5 | 0.31 | |
| P11 | 1.11 | 7.26 | 3.1 | 95 | 5.9 | 0.35 | |
| P21 | 1.11 | 6.75 | 3.1 | 95 | 5.9 | 0.35 | |
| P2 | 0.91 | 6.7 | 3.0 | 73 | 5.8 | 0.37 | |
| PEG | 1.27 | — | — | — | 42.6 | 0.34 | |
| NaCl | 1.53 | — | — | — | 200 | 1.15 | |
| Kinetic | k 0 | k 11 | k 21 | k 0°Cd | k 25°Cd | ||
| mL mol−1 min−1 | mL mol−1 min−1 | mL mol−1 min−1 | min−1 | min−1 | |||
| 22.8 | 17.9 | 24.0 | 0.003 | 0.01 | |||
 | ||
Fig. 3 Comparison between simulated on-column PEGylation chromatograms (solid lines) and experimental data (( ) P0, ( ) P0, ( ) P1 = P11 + P21 and ( ) P1 = P11 + P21 and ( ) P2). The error bars are computed from the standard deviations of two repetitions. ) P2). The error bars are computed from the standard deviations of two repetitions. | ||
These two peaks P11 and P21 were fractionated and further analyzed. Their degree of PEGylation was confirmed by SEC and mass spectrometry analysis. The MALDI-ToF scans of P1 (obtained by solution PEGylation), P11 and P21 are shown in Fig. 4 and confirm that these peaks correspond to mono-PEGylated α-lactalbumin. There is a slight deviation between P11 and P21 which could not be explained since both mono-PEGylated proteins are expected to have the same molecular weight which should also be identical to the one obtained for P1.
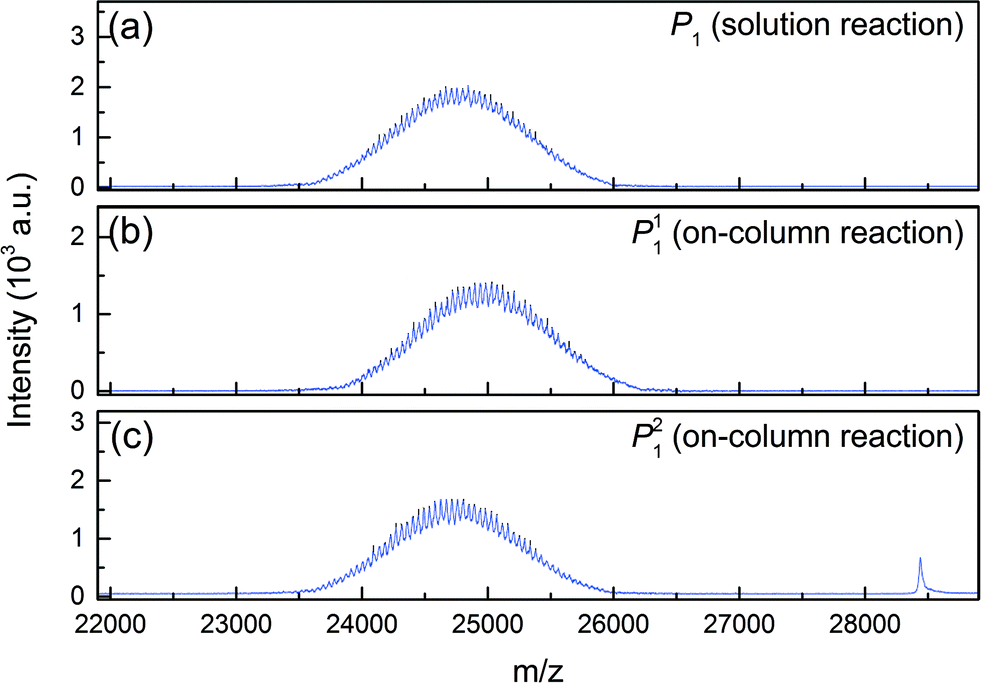 | ||
| Fig. 4 MALDI-ToF scans of mono-PEGylated α-lactalbumin obtained by (a) solution PEGylation and (b, c) on-column PEGylation at pH 7.0. | ||
As P1 was obtained in solution PEGylation, it is expected to be composed of P11 and P21, and therefore the MALDI-ToF scan of P1, shown in Fig. 4a, represents an average distribution of the molecular weight distributions of the two positional isomers. It is interesting to note that the polydispersity of the PEG polymer is reflected in the PEGylated product. Indeed, rather broad distributions are obtained and the mass difference between two consecutive peaks is 44 g mol−1, the mass of an ethylene glycol monomeric unit. The average molecular weight of these peaks is about 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 g mol−1 which corresponds to the molecular weight of α-lactalbumin (14178 g mol−1) conjugated with a PEG of 10500 g mol−1. This value is consistent with the supplier's data (10 kDa).
700 g mol−1 which corresponds to the molecular weight of α-lactalbumin (14178 g mol−1) conjugated with a PEG of 10500 g mol−1. This value is consistent with the supplier's data (10 kDa).
The design batch experiment
The feasibility of the internal recycling of the unreacted protein, by reversing the flow direction in the ‘elution’ column at the end of phase 4, had to be demonstrated. To do so, we developed the design batch experiment where a first reactive chromatography run was performed using the column in the ‘normal’ flow direction. At the end of the reaction, all the products but the unreacted protein were eluted. Subsequently, the column was turned, to apply the ‘reverse’ flow mode, and loaded with 1 mg of fresh protein. A second reactive chromatography run was performed. The same elution strategy was applied (i.e. the unreacted protein was kept adsorbed in the column). Finally, the remaining adsorbed protein was eluted by applying a high concentration of modifier in the elution buffer solution (1 M NaCl). The elution profiles together with the applied modifier concentration gradient and data from the analytics are shown in Fig. 5a. It can be seen that the method leads to very similar elution profiles in both flow directions. Indeed, the chromatograms look very similar and the different peaks elute at the same elution time in both flow directions. | ||
Fig. 5 (a) Chromatogram of the design batch experiment in normal flow direction (→), and in reverse flow (←), and complete elution of the remaining unreacted protein. (b) Comparison between simulated chromatograms (solid lines) and experimental data for the normal flow direction (→), and reverse flow direction (←). ( ) P0, ( ) P0, ( ) P1 = P11 + P21, ( ) P1 = P11 + P21, ( ) P2 and ( ) P2 and ( ) P3 (left axis) and the dashed lines correspond to the modifier concentration profile (right axis). ) P3 (left axis) and the dashed lines correspond to the modifier concentration profile (right axis). | ||
As previously observed, almost no tri-PEGylated protein was formed. Moreover, the concentrations of the different solutes in the second run are significantly higher than those in the first run. This shows that the unreacted protein indeed remains adsorbed inside the column, accumulates due to the additional 1 mg of protein loaded, and thus leads to a higher reactant concentration in the second run. Finally, the final stripping of the column showed a large peak identified as the remaining unreacted protein adsorbed on the resin at the end of the second PEGylation reaction.
The design batch experiment described above was simulated using the previously developed on-column PEGylation model. The model parameters for the chromatographic simulation as well as the kinetic rate constants of the on-column PEGylation reaction are given in Table 1. In Fig. 5b, the experimental results are compared with the result of the simulations for both runs. One can see that the quantities of products (multi- and mono-PEGylated α-lactalbumin and the unreacted protein), as well as their corresponding retention times and peak shapes, are correctly predicted by the model. This experiment not only proved the feasibility of the reverse flow switching method, it also confirmed the validity of the developed model. This model, together with the parameters given in Table 1, is further used to simulate the rMCSGP process.
rMCSGP operating parameters
The rMCSGP process uses the same stationary phase, the same columns as well as the same elution buffer solutions as the batch process. The design of the multi-column process can therefore be obtained directly from the batch elution experiments (see Fig. 5). The operating parameters for the MCSGP are obtained from the batch chromatogram as described by Müller-Späth et al.16 The slope of the modifier gradient applied in the rMCSGP process in the ‘elution’ column is the same as the one used in the design batch experiment.In order to assess the process performance, the yield, productivity and purity were defined. The process yield refers to the yield of mono-PEGylated α-lactalbumin at the process outlet, and is defined by:
 | (8) |
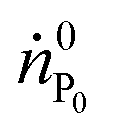 is the molar flow of protein at the process inlet. The product purity is defined as the ratio between the amount of the product in the outlet stream divided by the total solute molar flow (i.e. the sum of the molar flows of the unreacted protein and PEGylated proteins) in the same stream (ṅtot):
is the molar flow of protein at the process inlet. The product purity is defined as the ratio between the amount of the product in the outlet stream divided by the total solute molar flow (i.e. the sum of the molar flows of the unreacted protein and PEGylated proteins) in the same stream (ṅtot):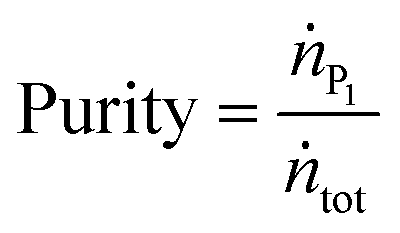 | (9) |
Finally, the productivity, in milligrams per hour equals the outlet molar flow times the molecular weight of the product (i.e. 24812 g mol−1 from the MALDI-ToF):
| Productivity = Mn,P1ṅP1 | (10) |
Operating constraints were set as follows: a purity constraint of 95% for the product, i.e. the mono-PEGylated α-lactalbumin, and a lower bound of 30% for the internally recycled streams. In other words, fractions with purity values below 30% were not recycled. Fig. 6 shows the chromatogram corresponding to the design batch experiment in the normal flow direction together with the estimated purity data. The purity data were obtained from the analytical SEC experiments. The dark grey area defines the pure product fraction collection window (purity above 95%) while the light grey areas define the recycled fractions (purity between 30 and 95%). It is also worth noting that the simulated purity curve is in really good agreement with the experimental data.
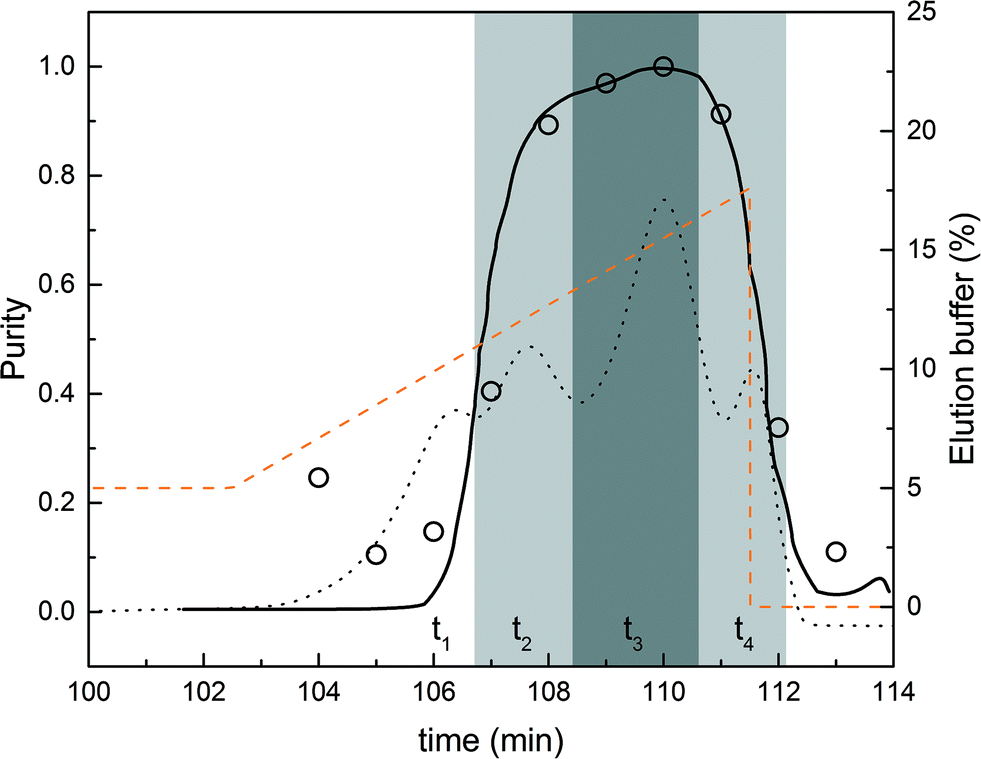 | ||
| Fig. 6 Comparison between experimental (○) and simulated (solid line) purity profiles of the mono-PEGylated protein (left axis). The collection and recycling intervals are shown as grey areas. The dark grey area is the collection windows (purity above 95%) while the light grey areas correspond to the recycling fractions. The modifier concentration profile is shown as a dashed line (right axis). The chromatogram corresponding to the design experiment in the normal flow direction is also presented (dashed line) for visual purposes. The scale of the chromatogram was adjusted to get a simplified representation and does not correspond to any axes. | ||
The times corresponding to the different phases are determined from Fig. 6. The interconnected times (t2 and t4) were estimated from the volumes to be recycled (corresponding to the two light grey areas). This procedure for estimating the operating parameters of the rMCSGP unit from a batch chromatogram so as to obtain the desired process performance is conceptually similar to the one originally developed for the non-reactive MCSGP10,11,15,16,18 and is shown here to work equally well. Nevertheless, the modifier gradient and interconnected times can later be fine-tuned to optimize the process performance and meet the desired purity and yield. In the rMCSGP process, the flow rates were also adapted in the ‘elution’ and ‘capture’ columns so that the elution time matches with the reaction time. The process parameters used for running the rMCSGP are summarized in Table 2, in which the different flow rates presented in Fig. 2 are given as well as the feed concentration of the reactants (functionalized PEG and protein). The variations with time of the process parameters for a complete switch time are reported.
| Time | Elution | Capture | Reaction | ||||
|---|---|---|---|---|---|---|---|
| Inlet flow mL min−1 | Buffer % | Inlet flow mL min−1 | Dilution % | Inlet flow mL min−1 | Feed concentration μmol L−1 | ||
| Min | |||||||
| t 1 | 0.00 | Q 1: 1.0 | 5.0 | 0 | 0.0 | Q 7: 1.0 | c 0,P0 = 28.2 |
| 2.50 | 5.0 | 0.0 | |||||
| 2.51 | 5.0 | 0.0 | Q 8: 0.03 | c 0,PEG = 451.2 | |||
| 10.00 | 5.0 | 0.0 | |||||
| 15.00 | 12.0 | 0.0 | |||||
| t 2 | 15.01 | Q 2: 0.1 | 12.0 | Q 2 + Q3: 1 | 90 | Q 8: 0.03 | |
| 37.25 | 14.1 | 90 | |||||
| t 3 | 37.26 | Q 4: 0.1 | 14.1 | 0 | 0.0 | Q 8: 0.03 | |
| 56.00 | 17.6 | 0.0 | |||||
| t 4 | 56.01 | Q 5: 0.1 | 0.0 | Q 5 + Q6: 1 | 90 | Q 8: 0.03 | |
| 81.00 | 0.0 | 90 | |||||
| 81.01 | 0.0 | 90 | Q 9: 1.0 | c 0,P0 = 0.0 | |||
| 95.00 | 0.0 | 90 | c 0,PEG = 0.0 | ||||
Reactive MCSGP simulations
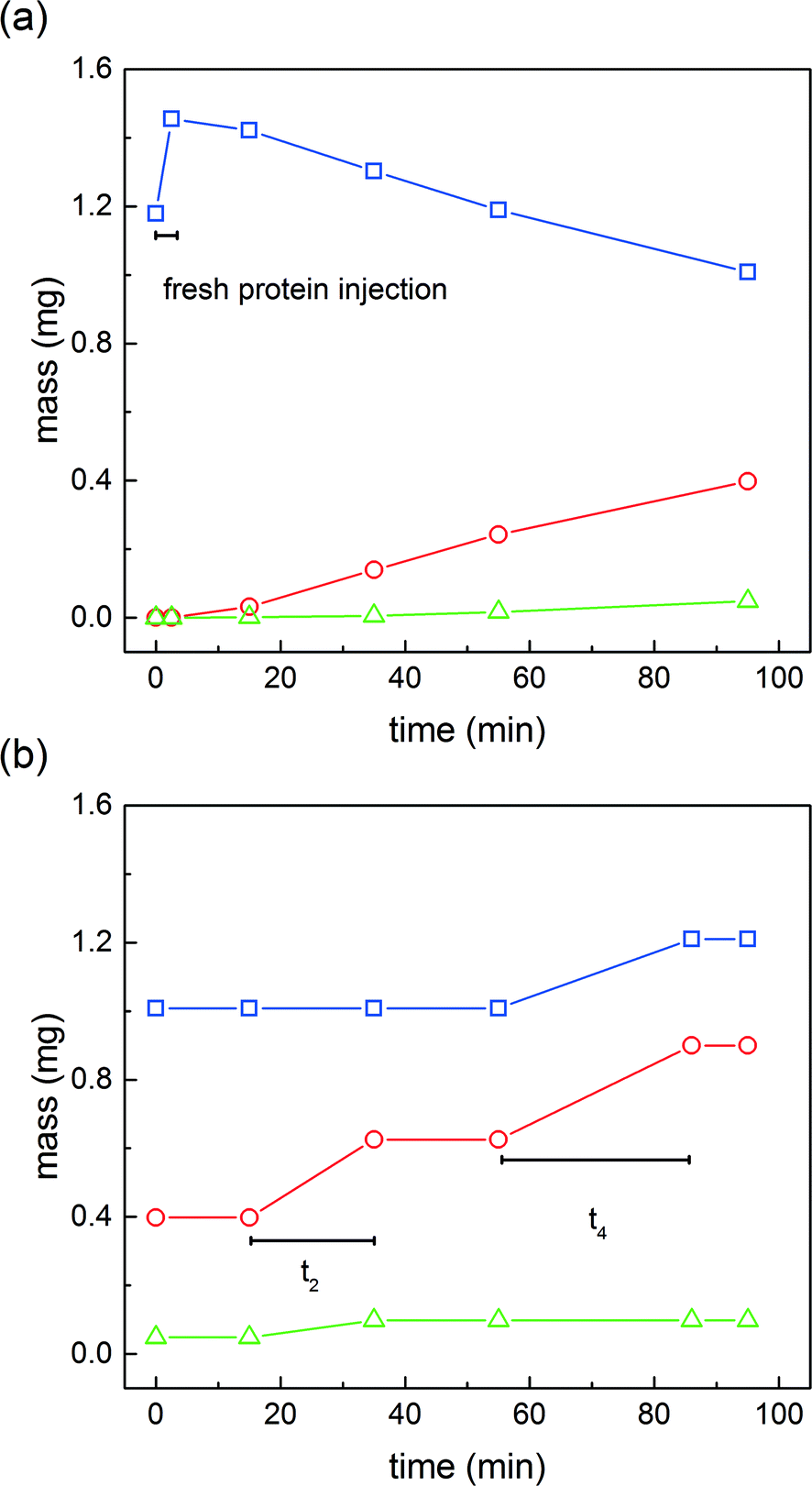 | ||
Fig. 7 Mass of native protein ( ), mono-PEGylated protein ( ), mono-PEGylated protein ( ) and multi-PEGylated proteins ( ) and multi-PEGylated proteins ( ) during one switch inside the ‘reaction’ column (a) and the ‘capture’ column (b). ) during one switch inside the ‘reaction’ column (a) and the ‘capture’ column (b). | ||
 | ||
| Fig. 8 Time evolution of the solid phase concentration profiles in the ‘elution’ column (P0, solid line, P1, dashed line and P2, dotted line). | ||
Fig. 7a shows the evolution of the amount of solutes in the ‘reaction’ column over time. It can be seen that after a short time of 2.5 minutes, the mass of protein increased from 1.12 to 1.45 mg. The difference of 0.33 mg corresponds to the quantity injected in the first phase of the reaction protocol (see Table 2). It is also worth noting that 1.12 mg is the mass of protein internally recycled at steady state. After this time, the quantity of α-lactalbumin decreases, the native protein being converted into mono-PEGylated and di-PEGylated proteins.
In the meantime, the ‘capture’ column is intermittently loaded with the content of the overlapping fractions coming from the ‘elution’ column. The time evolution of the quantity of products adsorbed in the ‘capture’ column is shown in Fig. 7b. At time 0, the quantities of solute adsorbed on the resin correspond to the quantities observed at the end of the on-column PEGylation reaction. Then, it can be observed that the mass of mono-PEGylated and di-PEGylated proteins increases over time t2 (22 minutes) corresponding to the first interconnected phase. Later, the quantities of mono-PEGylated protein and unreacted protein also increase over time t4 (40 minutes) corresponding to the second interconnected phase.
Additional insights can be gained by considering the internal column profiles at the end of the different phases of a switch. Fig. 8 shows, for the ‘elution’ column, the evolution of the internal solid phase concentration profiles over time. At time 0, the peculiar concentration profile is derived from the products of the reaction in addition to the recycled fractions. After a short time, the weakly adsorbing di-PEGylated protein is eluted under the effect of the modifier gradient.
This by-product is discarded. Once the mono-PEGylated product starts eluting (as shown in Fig. 8 after 15 minutes), the ‘elution’ column becomes connected to the ‘capture’ column in order to recycle this overlapping fraction. At the end of this phase, the two columns are disconnected and the product is collected at the desired purity. This is shown in Fig. 8 after 35 minutes. At the end of the product collection period, the fraction containing the product together with the unreacted protein is further recycled into the ‘capture’ column. Then, before the unreacted protein breaks through, the salt concentration in the elution buffer solution is strongly decreased so as to ‘trap’ the unreacted protein bound onto the resin of the ‘elution’ column. The corresponding concentration profile is shown in Fig. 8 at 95 minutes. The on-column PEGylation reaction protocol then starts from this configuration.
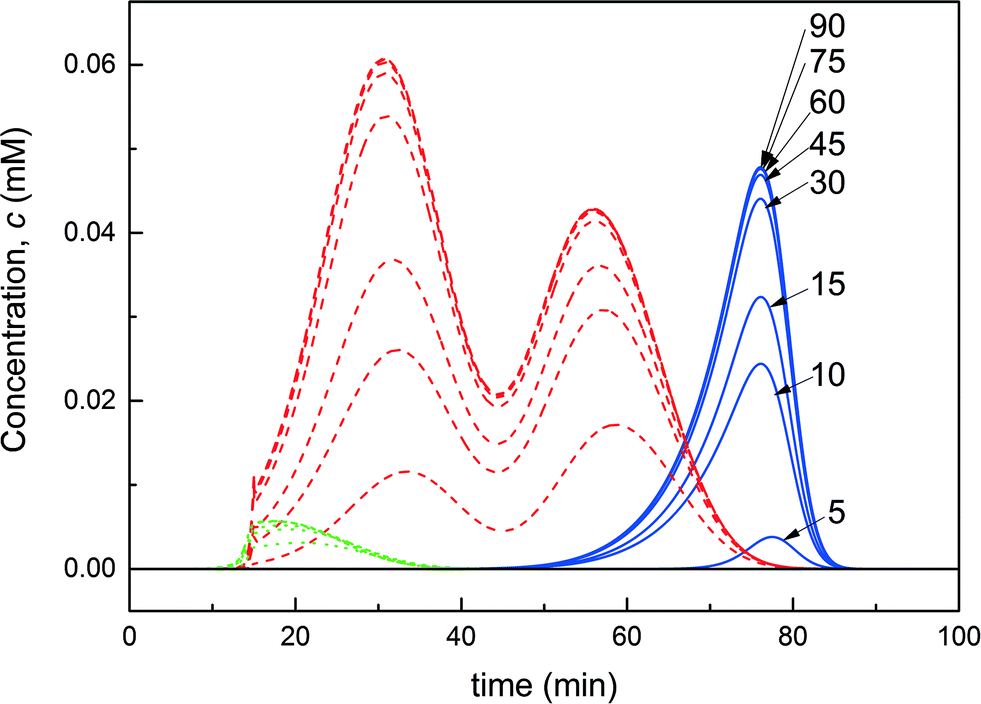 | ||
| Fig. 9 Overlay of the chromatograms obtained at the ‘elution’ column outlet representing the protein P0 (solid line), the product P1 (dashed line) and the multi-PEGylated protein (dotted) and shown cycle after cycle from the 5th to the 90th cycle. | ||
Fig. 10a shows the evolution of the process performance (purity, yield and productivity) as a function of the cycle number for the ‘base case scenario’. In the first two switches, all performance values are equal to 0, as no product elutes from the ‘elution’ column which is empty until the third cycle. From this cycle, until reaching steady state, the product purity remains greater than 95%. As already illustrated in Fig. 9, the convergence of the process is slow, and it is only after about 45 cycles that the process reaches steady state. The yield and the productivity reach a constant value of 80% and 0.9 mg h−1, respectively. The maximum possible productivity is dictated by the yield of the PEGylation reaction on the one hand and the yield of the separation on the other hand. Indeed, the formation of multi-PEGylated proteins negatively affects the productivity since a portion of the injected protein is wasted through this side reaction. Meanwhile, the product co-eluting with the multi-PEGylated protein in the early eluting fraction gets irremediably discarded. Therefore, if both reaction and separation were operated at the maximum yield, the productivity would reach 1.1 mg h−1. In our case, the process is operated at 80% of this maximum theoretical productivity.
 | ||
| Fig. 10 Purity, yield and productivity for the mono-PEGylated α-lactalbumin: (a) for the ‘base case scenario’ and (b) for the ‘transient acceleration’ strategy. | ||
In comparison, the productivity of the equivalent batch process without recycling the protein hardly reaches 0.3 mg h−1, which is about one third that of the rMCSGP process, for a yield of 44%. It has been shown in our previous article7 that the on-column PEGylation process presents the great advantage to be more selective for low degrees of PEGylation since the coupling reaction is restricted to fewer reaction sites compared to the solution reaction. Unfortunately, the performance of the on-column process was limited by the low conversion of the protein (between 25 and 55% depending on the experimental conditions) due to the decreased reaction rate which therefore led to a low productivity. The rMCSGP process, with its internal recycling of the unreacted protein, is shown to overcome this limitation since the conversion of the protein could be significantly increased. Moreover, the yield of the separation was also substantially improved thanks to the recycling of the non-resolved fractions. These two aspects explain the increased productivity and yield of the rMCSGP process compared to the batch process.
In order to shorten the transient to achieve steady state, a different start-up strategy was investigated. The process is expected to accumulate some unreacted protein before reaching steady state. In the previous example, in which 0.3 mg of fresh protein was injected into the ‘reaction’ column every run, the mass of internally recycled protein at steady state was 1.12 mg. Based on this observation, we tried to accelerate the process transient by feeding a larger amount of protein in the first cycle, and then loading 1 mg of protein per cycle as previously described. This ‘transient acceleration’ strategy was applied by injecting 10 mg of α-lactalbumin in the first cycle and then only 1 mg in the subsequent cycles. The evolution of the process performance cycle after cycle is shown in Fig. 10b. It can be seen that the steady state is reached after only 15 cycles, which is about three times faster than the ‘base case scenario’. Moreover, it is seen from the yield and productivity profiles that these parameters are oscillating before reaching steady state. The amplitude of the oscillation is limited (about 20% of the steady state value in the worst case) and reduces rapidly before reaching a constant value. Moreover, it is worth noting that the purity of the product, as well as all the other performance parameters at steady state, is not affected by the starting procedure, as expected.
Conclusion
The batch solution reaction of protein PEGylation at the lysine residues is known to exhibit a trade-off between the yield of the mono-PEGylated protein and conversion of the protein because of the further reactivity of the product of the reaction. In order to decouple the yield from the conversion, the integration of a chromatographic unit in the flowsheet, so as to isolate and recycle the unreacted protein, is a solution of choice. By doing so, the solution PEGylation can be performed in a batch reactor at a high yield of the mono-PEGylated product but at low conversion, the recycling enabling high conversion and high yield to be reached simultaneously. On the other hand, the on-column PEGylation process was shown to be a promising alternative to the classical batch solution reaction to improve the selectivity of the coupling reaction towards lower degrees of PEGylation. Indeed, the adsorption of the protein at the adsorbent surface leads to the reduction of the number of coupling sites because of steric hindrance. While the batch on-column PEGylation process has been reported in a previous work, the implementation of the on-column PEGylation in a multi-column continuous chromatographic process was investigated in this work. Here again, the idea was to decouple the yield from the conversion and therefore reach high process performance in a single unit. To this purpose, a multicolumn process in the form of a twin-column MCSGP unit was fitted with an additional column so that the on-column PEGylation reaction could be performed simultaneously with the purification of the products of the reaction.Based on an established method to design the MCSGP process, a reference batch on-column PEGylation experiment was performed and subsequently used as a benchmark to develop the reactive MCSGP setup. Compared to the classical MCSGP process, the rMCSGP process was modified in order to internally recycle the unreacted protein and therefore maximize the conversion of the protein. To do so, the elution of the protein was stopped just before it could leave the column and the flow direction was reversed in this column so that the protein was ready to react with the functionalized PEG in the next switch time.
Thanks to the results presented in our previous work, a mathematical model was derived and used to simulate this rMCSGP process. It was shown that, thanks to the internal recycling of non-resolved fractions and unreacted protein, the yield and the productivity for the mono-PEGylated α-lactalbumin were significantly increased compared to the batch process for the same purity of the mono-PEGylated protein. By imposing a purity of the product greater than 95%, a productivity of 0.9 mg h−1 of the product with a yield of 85% was obtained. The productivity of the rMCSGP process was shown to be far superior compared to the one obtained with the batch on-column process (0.3 mg h−1 at 44% yield).
The process was observed to reach steady state after about 45 cycles. The transient of the process was accelerated by implementing a ‘transient acceleration’ approach for which a larger amount of protein was loaded in the first cycle. By doing so, the time needed to reach steady state was divided by three.
It has been demonstrated that the integration of the PEGylation reaction and separation steps constitutes the foundation for the development of highly efficient PEGylation technologies.
Acknowledgements
This work was financially supported by Sanofi, Vitry sur Seine, France and by ETH Research Grant ETH-22 13-1. We would like to thank our colleague Daniel Baur for his precious help simulating the MCSGP process and his programming skills.References
- F. M. Veronese, A. Mero and G. Pasut, in PEGylated Protein Drugs: Basic Science and Clinical Applications, Springer, 2009, pp. 11–31 Search PubMed.
- D. Pfister and M. Morbidelli, J. Controlled Release, 2014, 180, 134–149 CrossRef CAS PubMed.
- D. Pfister, E. Bourgeaux and M. Morbidelli, Chem. Eng. Sci., 2015, 137, 816–827 CrossRef CAS.
- B. K. Lee, J. S. Kwon, H. J. Kim, S. Yamamoto and E. Lee, Bioconjugate Chem., 2007, 18, 1728–1734 CrossRef CAS PubMed.
- Z. F. Huang, G. H. Zhu, C. C. Sun, J. G. Zhang, Y. Zhang, Y. T. Zhang, C. H. Ye, X. J. Wang, D. Ilghari and X. K. Li, PLoS One, 2012, 7, e36423 CAS.
- X. Suo, X. Lu, T. Hu, G. Ma and Z. Su, Biotechnol. Lett., 2009, 31, 1191–1196 CrossRef CAS PubMed.
- D. Pfister, O. Ingold and M. Morbidelli, React. Chem. Eng., 2016 10.1039/C5RE00019J.
- A. Jungbauer, Trends Biotechnol., 2013, 31, 479–492 CrossRef CAS PubMed.
- L. Aumann and M. Morbidelli, Method and device for Chromatographic purification, Euro. pat., EP05405327.7, 2005 Search PubMed.
- L. Aumann and M. Morbidelli, Biotechnol. Bioeng., 2007, 98, 1043–1055 CrossRef CAS PubMed.
- L. Aumann, G. Stroehlein and M. Morbidelli, Biotechnol. Bioeng., 2007, 98, 1029–1042 CrossRef CAS PubMed.
- M. Krättli, T. Müller-Späth, N. Ulmer, G. Strohlein and M. Morbidelli, Ind. Eng. Chem. Res., 2013, 52, 8880–8886 CrossRef.
- R.-M. Nicoud, Ind. Eng. Chem. Res., 2014, 53, 3755–3765 CrossRef CAS.
- T. Müller-Späth, N. Ulmer, L. Aumann, C. Kennedy and M. Bavand, BioPharm Int., 2015, 28, 32 Search PubMed.
- T. Müller-Späth, N. Ulmer, L. Aumann, G. Strohlein, M. Bavand, L. J. A. Hendriks, J. de Kruif, M. Throsby and A. B. H. Bakker, BioProcess Int., 2013, 11, 36–45 Search PubMed.
- T. Müller-Späth, L. Aumann, L. Melter, G. Ströhlein and M. Morbidelli, Biotechnol. Bioeng., 2008, 100, 1166–1177 CrossRef PubMed.
- L. Aumann and M. Morbidelli, Biotechnol. Bioeng., 2008, 99, 728–733 CrossRef CAS PubMed.
- M. Krättli, T. Müller-Späth and M. Morbidelli, Biotechnol. Bioeng., 2013, 110, 2436–2444 CrossRef PubMed.
- R.-M. Nicoud, Chromatographic processes: modeling, simulation, and design, Cambridge University Press, Cambridge, 2015 Search PubMed.
- G. Carta and A. Jungbauer, Protein chromatography: process development and scale-up, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010 Search PubMed.
- M. Krättli, G. Stroehlein, L. Aumann, T. Muller-Spath and M. Morbidelli, J. Chromatogr. A, 2011, 1218, 9028–9036 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |