
A monolithic 5-(pyrrolidin-2-yl)tetrazole flow microreactor for the asymmetric aldol reaction in water–ethanol solvent†
Roberto
Greco
a,
Lorenzo
Caciolli
a,
Anna
Zaghi
a,
Omar
Pandoli
b,
Olga
Bortolini
a,
Alberto
Cavazzini
a,
Carmela
De Risi
a and
Alessandro
Massi
*a
aDipartimento di Scienze Chimiche e Farmaceutiche, Università di Ferrara, Via Fossato di Mortara 17, I-44121 Ferrara, Italy. E-mail: alessandro.massi@unife.it
bDepartamento de Química, Pontificia Universidade Católica – PUC-Rio, Rua Marques de São Vincente, 225, 22451-900, Rio de Janeiro, Brazil
First published on 4th November 2015
Abstract
An (S)-5-(pyrrolidin-2-yl)-1H-tetrazole organocatalyst has been prepared in the form of a monolithic column through the radical copolymerization of a styryl-functionalized pyrrolidinyl-tetrazole derivative, styrene and divinylbenzene in the presence of porogens (dodecanol and toluene). The activity of the monolithic pyrrolidinyl-tetrazole organocatalyst (triturated polymer) has been initially tested under batch conditions using the asymmetric aldol reaction of cyclohexanone and p-nitrobenzaldehyde as the benchmark. A prerequisite of the study has been the utilization of the eco-friendly water–ethanol mixture as the solvent. After having established the high efficiency and recyclability of the catalyst under these conditions, the effect of the flow regime has been evaluated by fabricating the corresponding monolithic microreactor (pressure-resistant stainless steel column). It has been demonstrated by a brief substrate scope study that the flow regime contributes to preserve the activity of the pyrrolidinyl-tetrazole catalyst over time (5 days on stream) with an almost twofold increase in productivity moving from batch to flow conditions. An added value of the flow procedure has been the optimization of a suitable 2D instrumental setup for simultaneous flow reaction and online flow-injection analysis.
Introduction
Continuous-flow meso- and micro-reactors can be considered nowadays useful scale-down versions of industrial reactors for transformations at the lab-scale;1 conversely, the scale-up of optimized microflow reactions is emerging as a new opportunity for the fine chemical industry with advantages in terms of safety, sustainability and ease of automation/monitoring of the production processes.2 In this scenario, continuous-flow asymmetric catalysis is receiving a great deal of attention as highlighted in recent reviews;3 indeed, utilization of microflow technologies as synthetic platforms for the production of multifunctional chiral molecules is very attractive, promising and worthy of investigation. Surprisingly, the implementation of organocatalyzed stereoselective flow processes has scarcely been considered until recently despite the peculiar benefits associated with the use of an organic molecule as a catalyst, which include stability in air and water, absence of metal leaching and compatibility with a number of unprotected functionalities. Very likely, this lack of initial interest had been directly related to the difficult recycling of organocatalysts and the high catalyst loadings often required for several transformations. Nevertheless, our group4 and others3a,b have recently demonstrated that heterogenization of organocatalysts may represent a viable solution to overcome the above drawbacks allowing the set-up of effective continuous-flow procedures endowed with high levels of stereoselectivity and productivity. The majority of the reported studies on asymmetric flow organocatalysis utilize the active catalyst immobilized on either polymeric or inorganic particles as a packing material for fixed-bed reactors. In view of an easy scale-up for potential industrial applications, this type of system, however, suffers from several limitations such as uncontrolled fluid dynamics, broad distribution of residence times, formation of hot-spots and stagnation zones.5 Monolithic reactors characterized by a regular or irregular network of meso- and micro-porous channels typically offer improved flow properties in virtue of the high void volume, which guarantees a large contact area of the catalyst with the fluid and the absence of a pressure drop.6 The immobilization of organocatalysts in the form of a continuous-flow monolithic columns is, however, still scant in the literature. Our group4c and that of Luis and García-Verdugo7 reported umpolung processes promoted by achiral azolium salts, while the group of Benaglia described stereoselective cycloadditions and Friedel–Crafts alkylations catalyzed by an imidazolidinone derivative.8As part of our ongoing studies on heterogeneous flow organocatalysis, we herein describe the fabrication of a polystyrene monolithic microreactor functionalized with the Ley–Arvidsson–Yamamoto (S)-5-(pyrrolidin-2-yl)-1H-tetrazole organocatalyst and its utilization in the stereoselective aldol reaction of cyclic ketones and acetone with activated aromatic aldehydes in the eco-friendly water–ethanol mixture. The asymmetric aldol reaction is one of the most versatile carbon–carbon bond-forming reactions leading to the widely diffused chiral β-hydroxyketone functionality.9 Continuous-flow heterogeneous organocatalysis has been applied to this fundamental reaction in a few studies using proline, proline mimetics and small peptides (Fig. 1);4d,e,10–14 each of the disclosed processes is characterized by strengths and limitations, although the procedure described by Pericàs and co-workers with a polystyrene immobilized proline derivative seems to excel in terms of diastereoselectivity of the aldol products.11
 | ||
| Fig. 1 Organocatalysts utilized for the continuous-flow asymmetric aldol reaction. | ||
The main goal of the present study is the disclosure of a monolithic microreactor with improved fluid dynamics and high long-term stability of the catalytic material for the effective production of chiral aldol products under environmentally benign conditions. A suitable 2D instrumental arrangement for simultaneous flow reaction and online flow-injection analysis has also been devised by coupling the pyrrolidinyl tetrazole-functionalized monolithic reactor to a chiral HPLC column for conversion and stereoselectivity determination.15
Results and discussion
In recent years, (S)-5-(pyrrolidin-2-yl)-1H-tetrazole has emerged as a ‘privileged’ organocatalyst that mediates a wide range of useful asymmetric reactions, including aldol, Michael, Mannich, α-amination reactions and so forth.16 In a previous study, we described a novel strategy based on photoinduced thiol–ene coupling (TEC) for the immobilization of a pyrrolidinyl-tetrazole catalyst on a silica support and demonstrated its activity in continuous-flow asymmetric aldol reactions.4d The use of low polarity solvents such as diisopropyl ether and toluene is found to be crucial to prevent the free hydroxyls on silica from perturbing the hydrogen-bonding network responsible for the high selectivities detected in the homogeneous catalysis. In the present study, we reversed our strategy and planned to create a favorable catalytic microenvironment in water medium by the occurrence of hydrophobic interactions between the polystyrene support and the catalytic sites through a simplified enzyme-like mechanism. Accordingly, the synthesis of the styryl pyrrolidinyl-tetrazole derivative 4 was initially addressed for its subsequent utilization as a co-monomer in the polymerization furnishing the target polystyrene monolithic column M6 (Schemes 1 and 2). Hence, the known styryl proline derivative 1 (ref. 17) was almost quantitatively converted into the amide 2 by in situ activation of the carboxylic acid. Subsequent dehydration of crude 2 promoted by cyanuric chloride afforded the nitrile 3 in good yield (75%), which in turn was transformed into the corresponding tetrazole 4 (72%) by cycloaddition with sodium azide (Scheme 1). | ||
| Scheme 1 Synthesis of the styryl-functionalized pyrrolidinyl-tetrazole 4. | ||
 | ||
| Scheme 2 Synthesis of polystyrene monolithic pyrrolidinyl-tetrazole M6. | ||
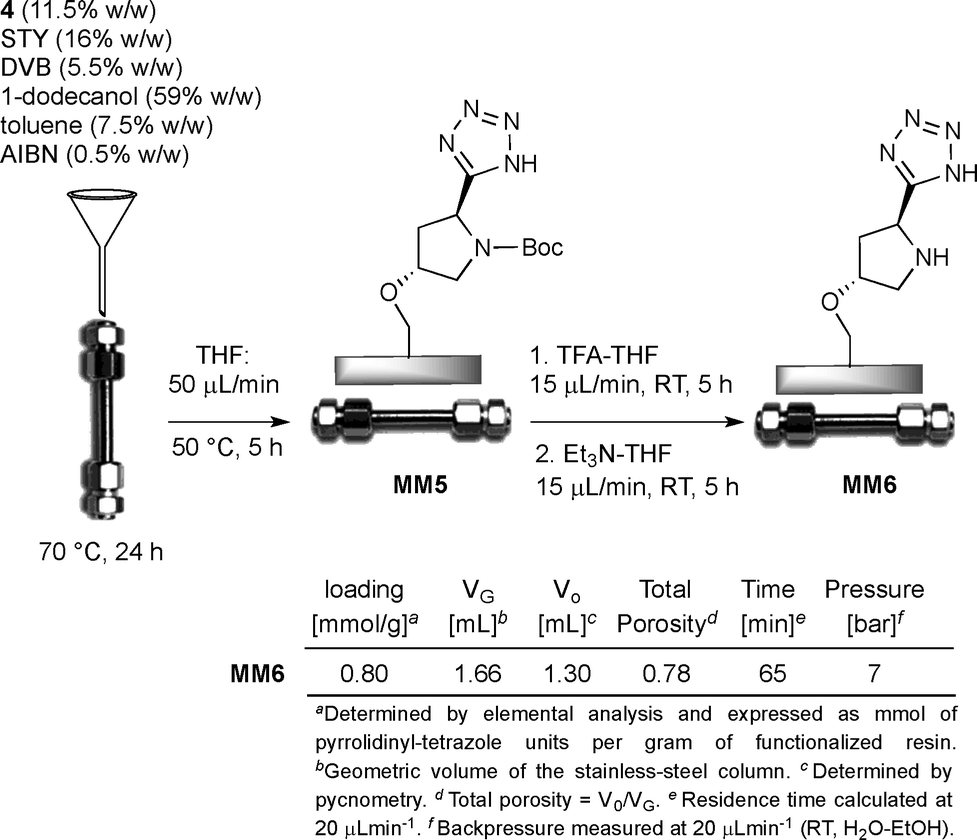
The synthesis of tetrazole-functionalized polystyrene monoliths was next investigated using the conditions first described by Fréchet and Svec.18 Therefore, an extensive polymerization study was conducted to identify the optimal ratio between styrene (STY), divinylbenzene (DVB), the co-monomer 4 and the porogens (toluene and 1-dodecanol). It is important to emphasize that monoliths were molded into both glass and stainless steel columns (4.6 mm internal diameter, 10 cm length) in parallel experiments to evaluate, on the one hand, the batch reactivity and the analytical/morphological properties of the prepared materials by SEM, FT-IR and elemental analysis; technically, the polymers prepared inside the glass columns were triturated and tested in the form of powder. On the other hand, this approach also allowed us to estimate the influence of the polymerization parameters on the flow-through behavior of the monolith inside the metal column since wall effects and high backpressures need to be carefully avoided for the correct functioning of the reactors in the flow regime. Hence, after some experimentation, the optimal composition of the polymerization mixture resulted as follows: 1-dodecanol (59% w/w), toluene (7.5% w/w), STY (16% w/w), DVB (5.5% w/w), 4 (11.5% w/w) and AIBN (0.5% w/w). Elemental analysis of the resulting monolith M5 indicated a satisfactory loading of pyrrolidinyl-tetrazole units (f: 0.82 mmol g−1); at the same time, 1H NMR analysis of the THF solution used to wash the polymer M5 confirmed the complete incorporation of the co-monomer 4 into the monolith. The N-Boc deprotection of triturated M5 was finally carried out with TFA/THF followed by treatment with Et3N/THF to give the target active catalyst M6. Several experiments were also attempted to directly polymerize the co-monomer 4 in its N-deprotected form; by this approach, however, the resulting monoliths M6 displayed detrimental pore clogging determining high backpressures in the flow regime and a very low reactivity under batch conditions, likely because of the reduced accessibility of the catalytic sites buried inside the micropores.
Preliminary batch experiments were performed with optimal M6 using the test reaction between cyclohexanone 7a and p-nitrobenzaldehyde 8a in different solvent systems, with particular attention to the environmentally benign water–ethanol mixture19 for the development of subsequent green applications of the newly prepared monolithic reactors (Table 1). Gratifyingly, the polystyrene pyrrolidinyl-tetrazole catalyst M6 was found to be highly active in pure water affording the aldol product 9a in quantitative yield and high stereoselectivity (anti/syn = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; 95% eeanti) in 4 h reaction time (entry 1). The addition of ethanol for achieving the full solubilization of the substrates (H2O/EtOH 1:1) determined a partial erosion of enantioselectivity (90% eeanti) accompanied by a marked decrease in diastereoselectivity (anti/syn = 5:1; entry 2). Unfortunately, and contrary to previous findings on the beneficial effect of temperature on the activity of pyrrolidinyl-tetrazole organocatalysts,4d,20 warming the reaction mixture to 50 °C produced a further drop in enantioselectivity (82% eeanti, entry 3). Next, a short solvent screening confirmed the necessary presence of water for obtaining satisfactory levels of conversion and stereoselectivity (entries 4–7). The maintenance of catalyst activity was finally ascertained in recycling experiments performed in H2O–EtOH, which was selected as the optimal solvent mixture for the subsequent flow experiments based on solubility reasons; catalyst M6 could be recycled up to eight times affording the aldol product 9a with an almost constant yield (turnover number TONbatch = 80) and only a moderate decrease in stereoselectivity in every cycle (entry 8). On the other hand, a peculiar advantage of the 1H-tetrazole moiety of M6 as isosteric replacement of the carboxylic acid functionality is the impossibility to undergo irreversible decarboxylation, which is a known side reaction occurring in the presence of activated aromatic aldehydes with analogue proline organocatalysts and leading to a progressive loss of catalytic activity.21
:1; 95% eeanti) in 4 h reaction time (entry 1). The addition of ethanol for achieving the full solubilization of the substrates (H2O/EtOH 1:1) determined a partial erosion of enantioselectivity (90% eeanti) accompanied by a marked decrease in diastereoselectivity (anti/syn = 5:1; entry 2). Unfortunately, and contrary to previous findings on the beneficial effect of temperature on the activity of pyrrolidinyl-tetrazole organocatalysts,4d,20 warming the reaction mixture to 50 °C produced a further drop in enantioselectivity (82% eeanti, entry 3). Next, a short solvent screening confirmed the necessary presence of water for obtaining satisfactory levels of conversion and stereoselectivity (entries 4–7). The maintenance of catalyst activity was finally ascertained in recycling experiments performed in H2O–EtOH, which was selected as the optimal solvent mixture for the subsequent flow experiments based on solubility reasons; catalyst M6 could be recycled up to eight times affording the aldol product 9a with an almost constant yield (turnover number TONbatch = 80) and only a moderate decrease in stereoselectivity in every cycle (entry 8). On the other hand, a peculiar advantage of the 1H-tetrazole moiety of M6 as isosteric replacement of the carboxylic acid functionality is the impossibility to undergo irreversible decarboxylation, which is a known side reaction occurring in the presence of activated aromatic aldehydes with analogue proline organocatalysts and leading to a progressive loss of catalytic activity.21
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Yieldb [%] | Time | d.r. anti/sync | eeantid [%] |
|
a Reactions performed in the stated solvent with 0.40 mmol of 8a (0.4 M) and 2.00 mmol of 7a.
b Isolated yield of the anti/syn diastereomeric mixture.
c Estimated by 1H NMR analysis of crude reaction mixtures.
d Determined by chiral HPLC analysis.
e 1:1 mixture.
f Reaction performed at 50 °C.
g 8th recycle.
|
|||||
| 1 | H2O | 95 | 4 | 10:1 |
95 |
| 2 | H2O–EtOHe | 95 | 4 | 5:1 |
90 |
| 3f | H2O–EtOHe | 95 | 1 | 4:1 |
82 |
| 4 | CH2Cl2 | nr | 24 | — | — |
| 5 | EtOH | 15 | 24 | 3:1 |
84 |
| 6 | DMF | 17 | 24 | 3:1 |
75 |
| 7 | H2O-DMFe | 85 | 4 | 8:1 |
90 |
| 8g | H2O–EtOHe | 95 | 4 | 5:1 |
88 |

At this stage of the study, we evaluated the performance of M6 as a monolithic microreactor (hereafter designed as MM). Thus, the optimized polymerization mixture containing the monomer 4 was transferred into the stainless steel column, which was then sealed at both ends and heated at 70 °C for 24 h in a standard convection oven (Scheme 3). After cooling, the resulting monolithic microreactor MM5 was connected to a HPLC instrument and then washed with THF (50 μL min−1; HPLC oven 50 °C, 5 h) to remove the porogen and residual non-polymeric material. The N-Boc deprotection step was next performed by sequentially flowing (15 μL min−1; 25 °C; 5 h) TFA/THF and Et3N/THF solutions through the column.
 | ||
| Scheme 3 Fabrication of monolithic microreactor MM6. | ||
The void volume and the total porosity of the monolithic microreactor MM6 were established by pycnometry. The main features of MM6 including the observed backpressure, residence time and catalyst loading are summarized in Scheme 3. The macroporous nature of MM6 was determined by SEM analysis (Fig. S1†); additionally, MM6 displayed nearly no swelling in water–ethanol (1:1) solvent as verified by dedicated experiments showing a linear relationship between backpressure and flow rate.22
As anticipated, integration of an analytical platform for monitoring the reaction progress in the flow regime was an important objective of this study. The devised injection–reaction–analysis system consisted in the monolithic microreactor MM6 connected to pump-1 through a mixer chamber (direction-1). The solutions of ketone 7 and aldehyde 8 were continuously fed into MM6 through channels A and B, respectively (Fig. 2). The outflow of the microreactor was redirected to a 6-port 2-position switching valve. In direction-2, the binary pump-2 delivered a hexane/i-PrOH solution into a chiral HPLC column by passing through the valve. This was controlled by a software program and allowed the switching from the ‘load’ position of the valve, where the sampling loop is filled with the effluent from MM6, to the ‘inject’ position, where the content of the loop is flushed into the chiral HPLC column for conversion and stereoselectivity determination. The developed 2D system greatly facilitated the study of the transition from batch to flow conditions of the model 7a/8a coupling, in which different flow rates and substrate concentrations were evaluated. Keeping in mind that reaction completion was a prior condition for an easier purification of the target aldol products, the optimal compromise between aldehyde solubility and process productivity (P) was found by injecting into MM6 a solution of p-nitrobenzaldehyde 7a (0.5 M) and cyclohexanone 8a (2.5 M) in H2O–EtOH (1:1.5) at 20 μL min−1 (residence time = 65 min) (Table 2, entry 1). Under these conditions, the steady-state regime was reached in ca. 4 h. Interestingly, the diastereoselectivity of the flow process (anti/syn = 7:1) slightly exceeded that of the batch reaction, while the enantioselectivity remained unmodified (90% eeanti). Isolation of the β-hydroxyketone 9a was simply achieved by extraction of the collected eluate with dichloromethane after 5 h operation of MM6 under steady-state conditions. Notably, the yield of isolated 9a was compared with that evaluated using the online analysis method, which resulted in a very similar value.
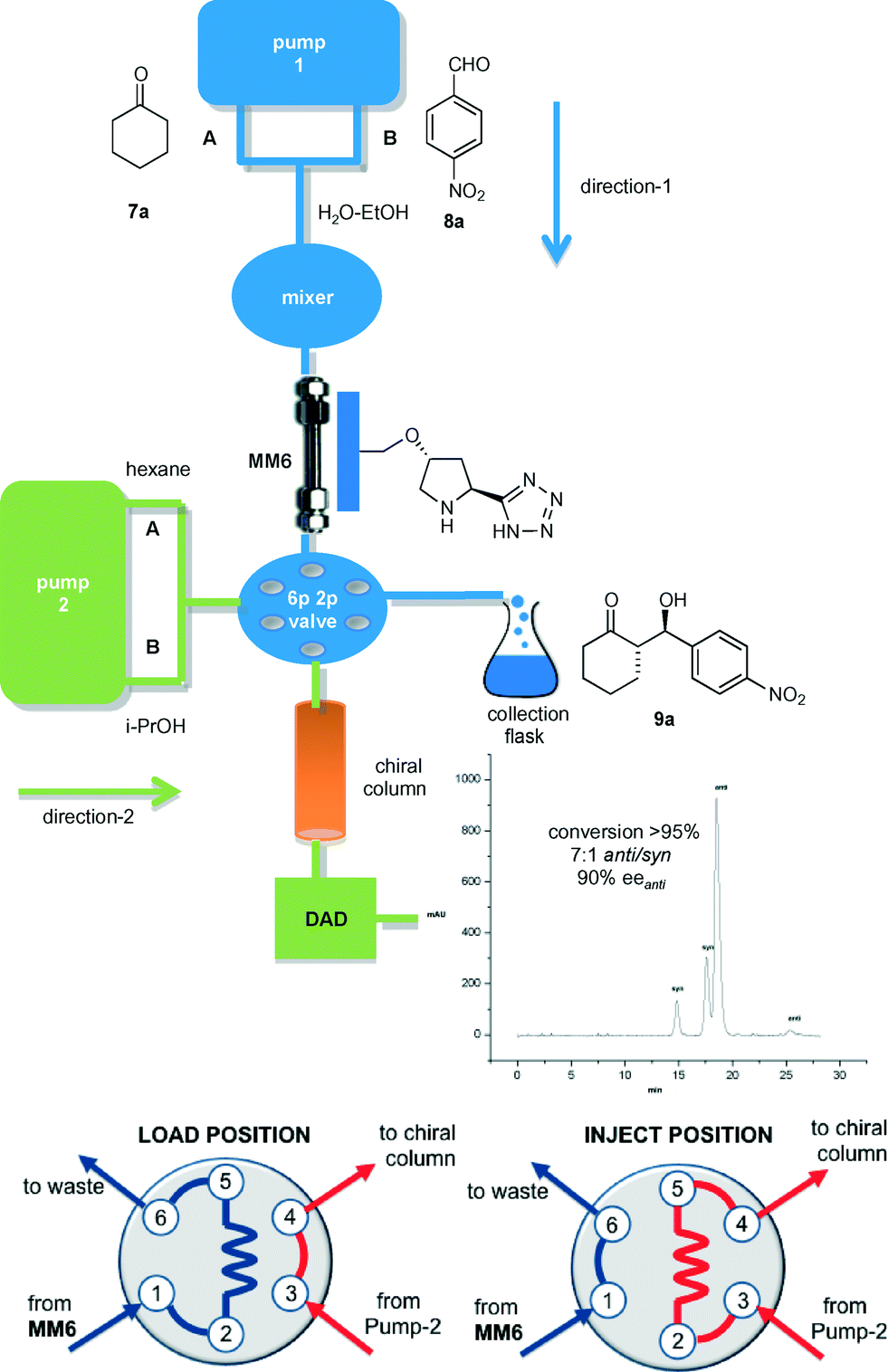 | ||
| Fig. 2 2D continuous-flow catalytic system for the production and online analysis of chiral β-hydroxyketones (6p2p valve: 6-port, 2-position switching valve; DAD: diode array detector). Magnification of the 6-port 2-position valve shows the ‘load’ and the ‘inject’ positions. | ||
| Entry | Products 9–11 | Flow rateb [μL min−1] | d.r. anti/sync | eeantid [%] | P |
|---|---|---|---|---|---|
|
a Experiments performed at 25 °C for 5 h (steady-state regime) in 1:1.5 H2O–EtOH ([7] = 2.5 M, [8] = 0.5 M).
b Flow rate required for the complete conversion of aldehyde 8.
c Estimated by 1H NMR analysis of the collected crude reaction mixtures.
d Determined by chiral HPLC analysis.
e
P (productivity) is measured in mmol (product) h−1 mmol (catalyst)−1.
f Experiments performed at 0 °C in 1.5:1 H2O–acetone ([8] = 0.5 M).
|
|||||
| 1 |
 9a
9a
|
20 | 7:1 |
90 | 1.22 |
| 2 |
 9b
9b
|
20 | 7:1 |
95 | 1.22 |
| 3 |
 9c
9c
|
15 | 6:1 |
89 | 0.92 |
| 4 |
 9d
9d
|
15 | 6:1 |
91 | 0.92 |
| 5 |
 9e
9e
|
20 | 5:1 |
95 | 1.22 |
| 6 |
 10a
10a
|
20 | 1:1 |
76 | 1.22 |
| 7 |
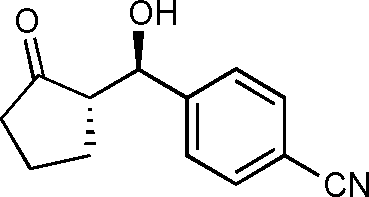 10b
10b
|
15 | 2:1 |
81 | 0.92 |
| 8 |
 10c
10c
|
15 | 1.5:1 |
71 | 0.92 |
| 9 |
 10d
10d
|
15 | 1.2:1 |
75 | 0.92 |
| 10 |
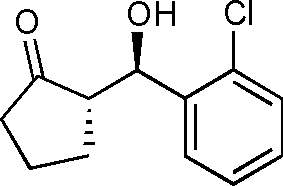 10e
10e
|
15 | 1:1 |
90 | 0.92 |
| 11f |
 11a
11a
|
15 | — | 81 | 0.92 |
| 12f |
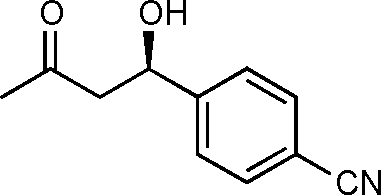 11b
11b
|
10 | — | 80 | 0.61 |
| 13f |
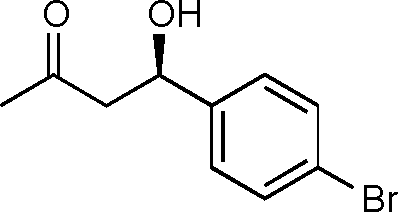 11c
11c
|
10 | — | 88 | 0.61 |
| 14f |
 11d
11d
|
10 | — | 71 | 0.61 |
| 15f |
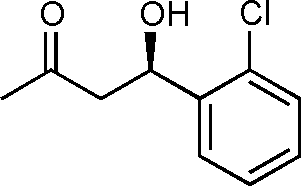 11e
11e
|
10 | — | 82 | 0.61 |
The long-term stability of monolithic microreactor MM6 was next examined to determine the effect of the flow regime on the deactivation rate of the heterogeneous pyrrolidinyl-tetrazole catalyst. The analysis of the reaction parameters versus process time plot shows that the steady-state conversion, diastereomeric excess (de) and ee are maintained unaltered for ca. 120 h on stream (TONflow = 147; Fig. 3). A progressive loss of efficiency is instead observed after that time, with a more pronounced effect at the level of reaction stereoselectivity.23
 | ||
| Fig. 3 Continuous-flow model aldol reaction in microreactor MM6 operated for 150 h. Conversion %, dashed line; de, plain line; ee, dotted line. | ||
Finally, the scope and applicability of the disclosed process and analysis method were briefly investigated in the aldol reactions of cyclohexanone 7a, cyclopentanone 7b or acetone 7c with the activated aromatic aldehydes 8a–e (Table 2). Gratifyingly, high conversion efficiencies could be achieved for all the substrate combinations by suitably adjusting the flow rate (10–20 μL min−1) of the optimized feed solutions. As for the model reaction, the monolithic column MM6 produced the aldol products 9b–e from cyclohexanone 7a with good enantioselectivity and satisfactory diastereoselectivity (entries 1–5). With cyclopentanone 7b as donor, the aldol products 10a–e were obtained as almost equimolar mixtures of diastereoisomers (entries 6–10). For the challenging aldol reaction of acetone in water medium, an acceptable level of enantioselectivity (71–88% ee) of the corresponding aldols 11a–e could be achieved by reducing the reaction temperature to 0 °C (entries 11–15).
Conclusions
In summary, we have presented a heterogeneous version of the valuable Ley–Arvidsson–Yamamoto catalyst in the form of a polystyrene monolithic column (MM6) and demonstrated its efficacy in the continuous production of optically active aldol products using the environmentally benign water–ethanol mixture as the reaction medium. A remarkable long-term stability of the catalytic bed (ca. five days on stream) could be achieved due to the resistance of the 1H-tetrazole moiety of MM6 toward known side reactions occurring with activated aldehydes as acceptor substrates.The devised 2D injection–reaction–analysis system together with the ease of product/catalyst separation and the detected twofold increase in productivity considerably enhanced the value of the optimized flow procedure compared to the corresponding batch protocol.
Monolithic columns of type MM6 can be fabricated at a low cost without the need for specialized equipment; an easy scale-up of the disclosed aldol process can be envisaged by the operation of multiple columns and the design of monoliths with larger diameters. Therefore, we hope that the methodology described herein may encourage further progress in the field of flow asymmetric catalysis and its utilization above the bench-scale.
Experimental section
All moisture-sensitive reactions were performed under a nitrogen atmosphere using oven-dried glassware. Solvents were dried over a standard drying agent and freshly distilled prior to use. Flash column chromatography was performed on silica gel 60 (230–400 mesh). Reactions were monitored by TLC on silica gel 60 F254 with detection by charring with sulfuric acid and/or phosphomolybdic acid. Optical rotations were measured at 20 ± 2 °C in the stated solvent; [α]D values are given in 10−1 deg cm2 g−1. 1H (300 MHz) and 13C (75 MHz) NMR spectra were recorded for CDCl3 solutions at room temperature unless otherwise specified. Peak assignments were aided by 1H–1H COSY and gradient-HMQC experiments. Enantiomeric excess (ee) values were determined by HPLC using the suitable chiral column (see below for details). Elemental analyses were performed using a FLASH 2000 Series CHNS/O analyzer (ThermoFisher Scientific). FT-IR analyses were performed using the Bruker Instrument Vertex 70. ESI-MS routine analyses were performed in positive ion mode with samples dissolved in 10 mM solution of ammonium formate in 1:1 MeCN/H2O. For accurate mass measurements, the compounds were analyzed in positive ion mode using an Agilent 6520 HPLC-Chip Q/TOF-MS (nanospray) with a quadrupole, a hexapole, and a time-of-flight unit to produce the spectra. The capillary source voltage was set at 1700 V; the gas temperature and drying gas were kept at 350 °C and 5 L min−1, respectively. The MS analyzer was externally calibrated with ESI-L low concentration tuning mix from m/z 118 to 2700 to yield accuracy below 5 ppm. Accurate mass data were collected by directly infusing samples in 40/60 H2O/ACN 0.1% TFA into the system at a flow rate of 0.4 μL min−1. SEM analyses were performed using a Zeiss Gemini 1530 scanning electron microscope. (2S,4R)-1-(tert-butoxycarbonyl)-4-((4-vinylbenzyl)oxy)pyrrolidine-2-carboxylic acid 1 (ref. 17) was synthesized as described. Adducts 9a–e, 10a–e and 11a–e are known compounds. The self-disproportionation of enantiomers (SDE) test for achiral chromatography24 has been carried out for derivatives 9a–e, 10a–e and 11a–e without noting any significant magnitude of SDE.
(2S,4R)-tert-Butyl 2-carbamoyl-4-((4-vinylbenzyl)oxy)pyrrolidine-1-carboxylate (2)
To a stirred mixture of (2S,4R)-1-(tert-butoxycarbonyl)-4-((4-vinylbenzyl)oxy)pyrrolidine-2-carboxylic acid 1 (ref. 17) (1.00 g, 2.88 mmol), di-tert-butyl dicarbonate (940 mg, 4.32 mmol), NH4HCO3 (341 mg, 4.32 mmol) and anhydrous CH3CN (15 mL), anhydrous pyridine (175 μL, 2.16 mmol) was added in one portion. The mixture was stirred at room temperature for 5 h, and then the volume was reduced under vacuum to approximately 5 mL. Subsequently, AcOEt (20 mL) and H2O (20 mL) were added, and the organic phase was separated. The aqueous phase was extracted further with AcOEt (2 × 20 mL), and the combined organic phases were washed with brine (10 mL), dried (Na2SO4), and concentrated to give the amide 2 (947 mg, 95%) at least 90% pure as determined by 1H NMR analysis. 1H NMR: δ = 7.40 (d, 2 H, J = 8.1 Hz, Ar), 7.32 (d, 2 H, J = 8.1 Hz, Ar), 7.28–7.20 (m, 2 H, Ar), 6.70 (dd, 1 H, J = 10.5 Hz, J = 17.0 Hz, HHC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 5.75 (d, 1 H, J = 17.0 Hz, HHCCH), 5.22 (d, 1 H, J = 10.5 Hz, HHCCH), 4.55–4.38 (m, 4 H, CH2O, H-2, H-4), 3.58–3.40 (m, 2 H, 2 H-5), 2.45–2.11 (m, 2 H, 2 H-3), 1.45 (s, 9 H, C(CH3)3); 13C NMR (two rotamers, selected data): δ = 175.9, 174.7, 156.8, 155.5, 137.6. 136.4, 127.8, 126.5, 114.5, 113.9, 80.7, 80.6, 76.8, 76.0, 70.9, 70.5, 59.6, 58.4, 52.0, 51.7, 37.6, 37.1, 28.3, 28.1. ESI MS (346.4): 347.8 (M + H+).
CH), 5.75 (d, 1 H, J = 17.0 Hz, HHCCH), 5.22 (d, 1 H, J = 10.5 Hz, HHCCH), 4.55–4.38 (m, 4 H, CH2O, H-2, H-4), 3.58–3.40 (m, 2 H, 2 H-5), 2.45–2.11 (m, 2 H, 2 H-3), 1.45 (s, 9 H, C(CH3)3); 13C NMR (two rotamers, selected data): δ = 175.9, 174.7, 156.8, 155.5, 137.6. 136.4, 127.8, 126.5, 114.5, 113.9, 80.7, 80.6, 76.8, 76.0, 70.9, 70.5, 59.6, 58.4, 52.0, 51.7, 37.6, 37.1, 28.3, 28.1. ESI MS (346.4): 347.8 (M + H+).
(2S,4R)-tert-Butyl 2-cyano-4-((4-vinylbenzyl)oxy)pyrrolidine-1-carboxylate (3)
To a cooled (0 °C), stirred solution of crude amide 2 (900 mg, 2.60 mmol) in anhydrous DMF (20 mL), cyanuric chloride (311 mg, 1.69 mmol) was added in one portion. The mixture was stirred at 0 °C for 1 h, then warmed to room temperature, and stirred at that temperature for an additional 24 h. The mixture was then cooled to 0 °C, diluted with H2O (15 mL) and extracted with AcOEt (3 × 50 mL). The combined organic phases were washed with brine (10 mL), dried (Na2SO4), concentrated and eluted from a column of silica gel with 3:1 cyclohexane–AcOEt to give 3 (640 mg, 75%) as a pale yellow oil. [α]D = −44.9 (c 1.4, CHCl3). 1H NMR: δ = 7.36 (d, 2 H, J = 8.1 Hz, Ar), 7.25 (d, 2 H, J = 8.1 Hz, Ar), 6.71 (dd, 1 H, J = 10.5 Hz, J = 17.5 Hz, HHCCH), 5.75 (d, 1 H, J = 17.5 Hz, HHCCH), 5.25 (d, 1 H, J = 10.5 Hz, HHCCH), 4.58–4.36 (m, 3 H, CH2O, H-2), 4.26–4.05 (m, 1 H, H-4), 3.77–3.37 (m, 2 H, 2 H-5), 2.53–2.21 (m, 2 H, 2 H-3), 1.51 (s, 9 H, C(CH3)3); 13C NMR (two rotamers, selected data): δ = 153.3, 137.8. 136.3, 128.2, 127.9, 126.4, 118.9, 114.2, 81.9, 75.2, 70.9, 50.7, 45.7, 37.4, 28.3. ESI MS (328.4): 329.6 (M + H+). HRMS (ESI) m/z calcd for C19H25N2O3 [M + H]+ 329.1865, found 329.1891.
(2S,4R)-tert-Butyl 2-(1H-tetrazol-5-yl)-4-((4-vinylbenzyl)oxy)pyrrolidine-1-carboxylate (4)
A mixture of nitrile 3 (700 mg, 2.13 mmol), NaN3 (181 mg, 2.78 mmol), Et3N·HCl (382 mg, 2.78 mmol), and toluene (10 mL) was stirred at 95 °C under an atmosphere of argon for 24 h. The mixture was then cooled to room temperature and extracted with AcOEt (3 × 40 mL). The combined organic phases were washed with brine (10 mL), dried (Na2SO4), concentrated, and eluted from a column of silica gel with 6:4:1 cyclohexane–AcOEt–AcOH to give 4 (569 mg, 72%) as a white amorphous solid. [α]D = −50.3 (c 1.0, CHCl3). 1H NMR: δ = 7.41 (d, 2 H, J = 8.1 Hz, Ar), 7.37 (d, 2 H, J = 8.1 Hz, Ar), 6.72 (dd, 1 H, J = 10.5 Hz, J = 17.5 Hz, HHCCH), 5.77 (d, 1 H, J = 17.5 Hz, HHCCH), 5.24 (d, 1 H, J = 10.5 Hz, HHCCH), 5.18 (t, 1 H, J = 7.3 Hz, H-2), 4.61–4.45 (m, 2 H, CH2O), 4.38–4.29 (m, 1 H, H-4), 3.65–3.58 (m, 1 H, H-5a), 3.45–3.37 (m, 1 H, H-5b), 3.10–2.96 (m, 1 H, H-3a), 2.65–2.50 (m, 1 H, H-3b), 1.49 (s, 9 H, C(CH3)3); 13C NMR (two rotamers, selected data): δ = 175.5, 156.5, 137.4, 136.3, 128.2, 127.9, 126.4, 114.2, 82.1, 76.2, 71.2, 52.1, 49.5, 34.5, 28.3. ESI MS (371.4): 372.5 (M + H+). HRMS (ESI) m/z calcd for C19H26N5O3 [M + H]+ 372.2036, found 372.2054.
Synthesis of monolithic pyrrolidinyl-tetrazole M6
A homogeneous mixture of tetrazole derivative 4 (180 mg, 0.49 mmol), styrene (280 μL, 2.43 mmol), divinylbenzene (technical grade, 80%; 100 μL, 0.70 mmol), toluene (140 μL, 1.31 mmol), 1-dodecanol (1.14 mL, 5.10 mmol), and AIBN (6 mg, 0.04 mmol) was degassed under vacuum and saturated with argon (by an Ar-filled balloon) three times. The mixture was then poured in a glass column (length: 10 cm, 0.46 cm internal diameter), sealed at both ends and placed in a vertical position in a standard convection oven. The polymerization was allowed to proceed for 24 h at 70 °C, then the column was cooled to room temperature, the glass was broken, and the resulting monolith M5 was triturated to obtain a yellow powder. This powder was suspended in THF (5 mL), centrifuged with 5 mL portions of THF (2 ×) and dried under reduced pressure (0.1 mbar, 40 °C, 6 h). The collected supernatants were analyzed (1H NMR analysis) to verify the complete incorporation of monomer 4 into the polymer. The triturated M5 resin was utilized for elemental and FT-IR analyses and the subsequent N-Boc deprotection step. Elemental analysis (%) found: N 5.74 (f = 0.82 mmol g−1). FT-IR (KBr): ν 3025, 2952, 1648, 1530 cm−1.To a cooled (0 °C), stirred mixture of triturated M5 resin and THF (0.8 mL), a solution of TFA (0.8 mL) and THF (0.8 mL) was slowly added. The mixture was then warmed to room temperature, stirred for 12 h, and centrifuged with 5 mL portions of THF (2×), 1:2 Et3N–THF (2×; addition at 0 °C) and THF (2×). The resulting monolith M6 was finally dried at a reduced pressure (0.1 mbar, 40 °C, 6 h). Elemental analysis (%) found: N 5.60 (f = 0.80 mmol g−1). FT-IR (KBr): ν 3022, 2950, 1521 cm−1.
Procedure for the model aldol reaction under batch conditions (Table 1)
A mixture of p-nitrobenzaldehyde (60 mg, 0.40 mmol), cyclohexanone (207 μL, 2.00 mmol), triturated monolith M6 (50 mg, 0.040 mmol) and the stated solvent (0.8 mL) was stirred at the stated temperature for the stated time and then centrifuged with 2.5 mL portions of CH2Cl2 (2×). The combined centrifugates were concentrated, and the resulting residue was analyzed by 1H NMR to determine the diastereomeric ratio and conversion. Subsequently, the residue was eluted from a column of silica gel with 5:1 toluene–AcOEt to determine the isolated yield of the anti/syn diastereomeric mixture and the enantiomeric excess value23 of the anti diastereoisomer by chiral HPLC analysis: Lux-1 cellulose (hexanes/i-PrOH 98:2 v/v, 400 μL min−1; λmax = 258 nm); tR (major) = 18.5 min; tR (minor) = 25.4.
Preparation of monolithic microreactor (MM6)
A homogeneous mixture of tetrazole derivative 4 (180 mg, 0.49 mmol), styrene (280 μL, 2.43 mmol), divinylbenzene (technical grade, 80%; 100 μL, 0.70 mmol), toluene (140 μL, 1.31 mmol), 1-dodecanol (1.14 mL, 5.10 mmol), and AIBN (6 mg, 0.04 mmol) was degassed under vacuum and saturated with argon (by an Ar-filled balloon) three times. The mixture was then poured in a stainless steel column (length: 10 cm, 0.46 cm internal diameter) sealed at both ends and placed in a vertical position in a standard convection oven. The polymerization was allowed to proceed for 24 h at 70 °C, then the column MM5 was cooled to room temperature and the seals were removed. The column was provided with fittings, connected to the HPLC pump, and warmed to 50 °C. Then, THF was pumped through the warmed column at a flow rate of 50 μL min−1 for 5 h to remove the porogenic solvents and residual non-polymeric materials. The column MM5 was cooled to room temperature; then, it was pumped with an input stream of 1:2 TFA–THF solution (5 mL) at 15 μL min−1 for 5 h followed by an input stream of 1:2 Et3N–THF solution (5 mL; 15 μL min−1 for 5 h). The resulting column MM6 was finally washed at room temperature with THF at a flow rate of 50 μL min−1 for 2 h. The whole procedure was performed with a reference microreactor in a parallel experiment with the aim of establishing the main feature of MM6 (level of functionalization, the void volume V0, the total porosity, the residence time and the observed backpressure; see Scheme 3).
Determination of the microreactor void volume
The microreactor void volume (V0) was determined by pycnometry.25 This method consists in filling the microreactor successively with two distinct solvents (solvent 1: water; solvent 2: n-hexane) and weighing the filled microreactors accurately. Simple math shows that:26| V0 = (ω1 − ω2)/(δ1 − δ2) |
Description of the experimental set-up for simultaneous flow-reaction and flow-injection analysis (Fig. 2 and Table 2)
The system used for simultaneous flow-reaction and flow-injection analysis was made up of two binary pumps (Agilent 1100 and Agilent 1100 micro series), a thermostated microreactor holder (Peltier unit), a diode array detector (DAD), and a data acquisition system (Agilent ChemStation).4b On the first direction, pump-1 was connected to the microreactor MM6 through a mixer chamber (200 μL). Channel-A was used to deliver a solution of ketone 7 (3.75 M) in H2O–EtOH (1:1.5). Channel-B delivered a solution of aldehyde 8 (0.75 M) in H2O–EtOH (1:1.5). The feed solutions were pumped at room temperature and at the stated flow rate through the mixer into MM6 by operating channel-A and channel-B of pump-1 at a 2:1 ratio. Experiments with acetone 7c as donor were performed at 0 °C in 1.5:1 H2O–acetone ([8] = 0.5 M). The effluent from MM6 was redirected to a 6-port 2-position switching valve. On the second direction, the binary pump-2 delivered a solution of hexanes/i-PrOH (see below for compositions) into the suitable chiral HPLC column by passing through the switching valve. The 6-port 2-position valve (Rheodyne) was controlled by a software program27 and allowed the switching between the ‘load’ and ‘inject’ positions for conversion and stereoselectivity determination. Microreactor MM6 was operated for 5 h under steady-state conditions, and then the collected solution was extracted with CH2Cl2 (ca. 20 min for the separation of the phases) to give the target β-hydroxyketone 9 as diastereomeric mixture at least 90% pure as determined by 1H NMR analysis.
The long-term stability experiment was performed using cyclohexanone 7a (0.25 M) and p-nitrobenzaldehyde 8a (0.5 M) as the substrates; microreactor MM6 was operated at 25 °C with a flow rate of 20 μL min−1 for ca. 150 h. After the achievement of the steady-state regime (ca. 4 h), full conversion of 7a was maintained for ca. 120 h, while a progressive loss of catalytic activity was observed after that time (TONflow = 147).
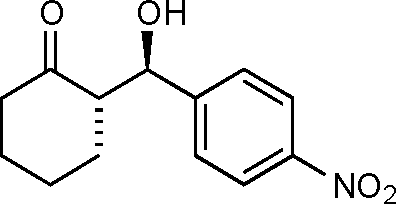
(S)-2-((R)-Hydroxy(4-nitrophenyl)methyl)cyclohexanone (9a)
Yield: 98% (0.73 g). 1H NMR: δ = 8.23 (d, 2 H, J = 8.7 Hz, Ar), 7.53 (d, 2 H, J = 8.7 Hz, Ar), 4.92 (dd, 1 H, J = 3.2 Hz, J = 8.4 Hz, H-1′), 4.10 (d, J = 3.2 Hz, OH), 2.38–2.62 (m, 3 H), 2.11–2.16 (m, 1 H), 1.38–1.87 (m, 5 H); 13C NMR: δ = 214.8, 148.4, 147.6, 127.9, 123.6, 74.0, 57.2, 42.7, 30.8, 27.7, 24.7; HPLC: Lux-1 cellulose (hexanes/i-PrOH 98:2 v/v, 0.4 mL min−1; λmax = 258 nm); tR (major) = 18.5 min; tR (minor) = 25.4.
(S)-2-((R)-Hydroxy(4-cyanophenyl)methyl)cyclohexanone (9b)
Yield: 97% (0.67 g). 1H NMR: δ =7.64 (d, 2 H, J = 8.4 Hz, Ar), 7.45 (d, 2 H, J = 8.4 Hz, Ar), 4.84 (d, 1 H, J = 8.4 Hz, H-1′), 2.54–2.61 (m, 1 H), 1.26–2.51 (m, 8 H). 13C NMR: δ = 214.8, 146.3, 132.1, 127.7, 118.7, 111.6, 74.1, 57.1, 42.6, 30.7, 27.6, 24.6. HPLC: Lux-1 cellulose (hexanes/i-PrOH 95:5 v/v, 1 mL min−1; λmax = 220 nm); tR (major) = 13.9 min; tR (minor) = 18.0.
(S)-2-((R)-Hydroxy(4-bromophenyl)methyl)cyclohexanone (9c)
Yield: 99% (0.63 g). 1H NMR: δ = 7.51–7.38 (m, 2 H, Ar), 7.24–7.12 (m, 2 H, Ar), 4.74 (d, 1 H, J = 8.7 Hz, H-1′), 4.03 (bs, 1 H, OH), 2.63–2.25 (m, 3 H), 2.15–1.94 (m, 1 H), 1.93–1.15 (m, 5 H). 13C NMR: δ = 215.3, 140.0, 131.5, 128.7, 121.7, 74.2, 57.3, 42.6, 30.7, 27.7, 24.7. HPLC: Lux-1 cellulose (hexanes/i-PrOH 95:5 v/v, 1 mL min−1; λmax = 220 nm); tR (major) = 13.9 min; tR (minor) = 18.0.
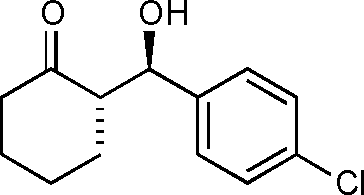
(S)-2-((R)-Hydroxy(4-chlorophenyl)methyl)cyclohexanone (9d)
Yield: 98% (0.52 g). 1H NMR: δ = 7.31 (d, 2 H, J = 8.4 Hz, Ar), 7.25 (d, 2 H, J = 8.6 Hz, Ar), 4.76 (d, 1 H, J = 8.8 Hz, H-1′), 2.58–2.43 (m, 2 H), 2.38–2.30 (m, 1 H), 2.11–2.04 (m, 1 H), 1.86–1.75 (m, 1 H), 1.72–1.48 (m, 3 H), 1.33–1.26 (m, 1 H). 13C NMR: δ = 215.4, 139.4, 133.6, 128.5, 128.4, 74.1, 57.3, 42.7, 30.7, 27.7, 24.7. HPLC: Lux-1 cellulose (hexanes/i-PrOH 99:1 v/v, 1 mL min−1; λmax = 220 nm); tR (major) = 29.4 min; tR (minor) = 40.0.
(S)-2-((R)-Hydroxy(2-chlorophenyl)methyl)cyclohexanone (9e)
Yield: 97% (0.69 g). 1H NMR: δ = 7.60–7.50 (m, 1 H, Ar), 7.38–7.15 (m, 3 H, Ar), 5.34 (dd, 1 H J = 8.1, Hz, J = 3.9 Hz, H-1′), 4.03 (d, 1 H, J = 3.9 Hz, OH), 2.72–2.63 (m, 1 H), 2.48–2.28 (m, 2 H), 2.11–2.05 (m, 1 H), 1.86–1.46 (m, 5 H). 13C NMR: δ = 215.3, 139.0, 132.9, 129.2, 128.7, 128.2, 127.2, 70.4, 57.6, 42.7, 30.4, 27.8, 24.9. HPLC: Lux-1 cellulose (hexanes/i-PrOH 99:1 v/v, 1 mL min−1; λmax = 220 nm); tR (major) = 20.7 min; tR (minor) = 25.5.
(S)-2-((R)-Hydroxy(4-nitrophenyl)methyl)cyclopentanone (10a)
Yield: 96% (0.68 g). 1H NMR: δ = 8.17–8.13 (m, 2 H, Ar), 7.49–7.45 (m, 2 H, Ar), 4.78 (d, 1 H, J = 9.2 Hz, H-1′), 4.69 (bs, 1 H, OH), 2.44–2.16 (m, 3 H), 2.00–1.90 (m, 1 H), 1.73–1.45 (m, 3 H); 13C NMR: δ = 222.0, 148.5, 147.5, 127.2, 123.6, 74.4, 55.1, 38.6, 26.9, 20.4. HPLC: Chiralpak IC (hexanes/i-PrOH 80:20 v/v, 0.5 mL min−1; λmax = 220 nm); tR (major) = 26.8 min; tR (minor) = 31.9.
(S)-2-((R)-Hydroxy(4-cyanophenyl)methyl)cyclopentanone (10b)
Yield: 97% (0.47 g). 1H NMR: δ = 7.66–7.60 (m, 2 H, Ar), 7.48–7.40 (m, 2 H, Ar), 4.79 (d, 1 H J = 9.2 Hz, H-1′), 4.72 (s, 1 H, OH), 2.50–1.50 (m, 7 H); 13C NMR: δ = 222.2, 148.3, 132.2, 127.2, 118.7, 111.7, 70.5, 55.0, 38.5, 26.7, 20.3. HPLC: Chiralpak ID (hexanes/i-PrOH 90:10 v/v, 1 mL min−1; λmax = 220 nm); tR (minor) = 30.8 min; tR (major) = 32.7.
(S)-2-((R)-Hydroxy(4-bromophenyl)methyl)cyclopentanone (10c)
Yield: 99% (0.60 g). 1H NMR: δ = 7.48 (d, 2 H, J = 8.4 Hz, Ar), 7.22 (d, 2 H, J = 8.4 Hz, Ar), 4.68 (d, 1 H, J = 8.9 Hz, H-1′), 2.36–1.89 (m, 5 H), 1.75–1.62 (m, 2 H); 13C NMR: δ = 222.8, 140.7, 131.5, 128.8, 121.9, 74.7, 55.5, 38.9, 26.9, 20.5. HPLC: Chiralpak ID (hexanes/i-PrOH 80:20 v/v, 1 mL min−1; λmax = 220 nm); tR (minor) = 8.4 min; tR (major) = 8.8.
(S)-2-((R)-Hydroxy(4-chlorophenyl)methyl)cyclopentanone (10d)
Yield: 97% (0.49 g). 1H NMR: δ = 7.35–7.21 (m, 4 H, Ar), 4.68 (d, 1 H, J = 9.0 Hz, H-1′), 4.62 (bs, 1 H, OH), 2.45–1.40 (m, 7 H); 13C NMR: δ = 222.4, 140.3, 133.5, 128.1, 128.6, 74.8, 55.3, 38.3, 27.0, 20.1. HPLC: Chiralpak ID (hexanes/i-PrOH 90:10 v/v, 1 mL min−1; λmax = 220 nm); tR (minor) = 13.5 min; tR (major) = 14.5.
(S)-2-((R)-Hydroxy(2-chlorophenyl)methyl)cyclopentanone (10e)
Yield: 98% (0.49 g). 1H NMR: δ = 7.59 (d, 1 H, J = 7.6 Hz, Ar), 7.45–7.27 (m, 3 H, Ar), 5.30 (d, 1 H, J = 9.2 Hz, H-1′), 4.52 (s, 1 H, OH), 2.28–2.52 (m, 3 H), 2.03–2.07 (m, 1 H), 1.71–1.80 (m, 3 H); 13C NMR: δ = 222.8, 139.1, 132.2, 129.1, 128.7, 128.2, 127.1, 70.2, 55.5, 38.6, 26.2, 20.4. HPLC: Chiralpak ID (hexanes/i-PrOH 95:5 v/v, 1 mL min−1; λmax = 220 nm); tR (major) = 33.2 min; tR (minor) = 38.2.
(R)-4-Hydroxy-4-(4-nitrophenyl)butan-2-one (11a)
Yield: 98% (0.31 g). 1H NMR: δ = 8.20 (d, 2 H, J = 8.8 Hz, Ar), 7.53 (d, 2 H, J = 8.8 Hz, Ar), 5–31–5.25 (m, 1 H, H-4), 3.57 (d, 1 H, J = 3.3 Hz, OH), 2.88–2.82 (m, 2 H), 2.22 (s, 3 H). 13C NMR: δ = 208.5, 149.9, 126.4, 123.8, 68.9, 51.5, 30.7. HPLC: Chiralpak ID (hexanes/i-PrOH 80:20 v/v, 0.5 mL min−1; λmax = 220 nm); tR (major) = 19.7 min; tR (minor) = 21.2.(R)-4-Hydroxy-4-(4-cyanophenyl)butan-2-one (11b)
Yield: 96% (0.27 g). 1H NMR: δ = 7.64 (d, 2 H, J = 8.0 Hz, Ar), 7.48 (d, 2 H, J = 8.0 Hz, Ar), 5.45–5.38 (m, 1 H, H-4), 2.70–2.55 (m, 2 H), 2.21 (s, 3H). 13C NMR: δ = 208.3, 148.1, 132.2, 126.3, 118.6, 111.2, 69.0, 51.5, 30.6. HPLC: Chiralpak ID (hexanes/i-PrOH 85:15 v/v, 1 mL min−1; λmax = 280 nm); tR (major) = 37.0 min; tR (minor) = 40.2.
(R)-4-Hydroxy-4-(4-bromophenyl)butan-2-one (11c)
Yield: 98% (0.35 g). 1H NMR: δ = 7.47 (d, 2 H, J = 8.4 Hz, Ar), 7.23 (d, 2 H, J = 8.0 Hz, Ar), 5.11 (q, 1 H, J = 4.0 Hz, J = 8.0 Hz, H-4), 2.83–2.81 (m, 2 H), 2.20 (s, 3 H); 13C NMR: δ = 208.9, 141.6, 131.4, 127.2, 121.2, 69.0, 51.6, 30.7. HPLC: Chiralpak ID (hexanes/i-PrOH 80:20 v/v, 1 mL min−1; λmax = 280 nm); tR (major) = 9.1 min; tR (minor) = 9.8.
(R)-4-Hydroxy-4-(4-chlorophenyl)butan-2-one (11d)
Yield: 97% (0.29 g). 1H NMR: δ = 7.28-7.33 (m, 4 H, Ar), 5.11–5.15 (q, 1 H, J = 4.0 Hz, J = 8.0 Hz, H-4), 2.88–2.78 (m, 2 H), 2.20 (s, 3 H); 13C NMR: δ = 208.9, 141.1, 133.0, 128.4, 126.9, 68.9, 51.6, 30.6. HPLC: Chiralpak ID (hexanes/i-PrOH 80:20 v/v, 1 mL min−1; λmax = 220 nm); tR (major) = 7.8 min; tR (minor) = 9.4.
(R)-4-Hydroxy-4-(2-chlorophenyl)butan-2-one (11e)
Yield: 98% (0.29 g). 1H NMR: δ = 7.63–7.61 (m, 1 H, Ar), 7.34–7.20 (m, 3 H, Ar), 5.52–5.48 (m, 1 H, H-4), 3.58 (d, 1 H, J = 3.2 Hz, OH), 3.05–2.60 (m, 2 H), 2.22 (s, 3 H). 13C NMR: δ = 208.9, 140.0, 130.7, 129.0, 128.3, 127.0, 126.8, 66.2, 49.9, 30.3. HPLC: Chiralpak ID (hexanes/i-PrOH 95:5 v/v, 0.5 mL min−1; λmax = 220 nm); tR (major) = 11.8 min; tR (minor) = 12.9.
Acknowledgements
We gratefully acknowledge the University of Ferrara (fondi FAR) for financial support. Thanks are also given to Mr Paolo Formaglio for NMR experiments, Mrs Tatiana Bernardi for HRMS analyses, and Mrs Ercolina Bianchini for elemental analyses.Notes and references
- (a) R. Munirathinam, J. Huskens and W. Verboom, Adv. Synth. Catal., 2015, 357, 1093–1123 CrossRef CAS; (b) D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed; (c) T. Wirth, Microreactors in Organic Chemistry and Catalysis, 2nd edn., Wiley-VCH, Weinheim, 2013 Search PubMed; (d) S. V. Luis and E. García-Verdugo, Chemical Reactions and Processes under Flow Conditions, RSC Publishing, Cambridge, 2009 Search PubMed.
- (a) J. C. Pastre, D. L. Browne and S. V. Ley, Chem. Soc. Rev., 2013, 42, 8849–8869 RSC; (b) V. Hessel, D. Kralisch, N. Kockmann, T. Noel and Q. Wang, ChemSusChem, 2013, 6, 746–789 CrossRef CAS PubMed; (c) I. R. Baxendale, L. Brocken and C. J. Mallia, Green Process. Synth., 2013, 2, 211–230 CAS; (d) J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS.
- (a) F. G. Finelli, L. S. M. Miranda and R. O. M. A. de Souza, Chem. Commun., 2015, 51, 3708–3722 RSC; (b) I. Atodiresei, C. Vila and M. Rueping, ACS Catal., 2015, 5, 1972–1985 CrossRef CAS; (c) A. Puglisi, M. Benaglia and V. Chiroli, Green Chem., 2013, 15, 1790–1813 RSC; (d) D. Zhao and K. Ding, ACS Catal., 2013, 3, 928–944 CrossRef CAS; (e) T. Tsubogo, T. Ishiwata and S. Kobayashi, Angew. Chem., Int. Ed., 2013, 52, 6590–6604 CrossRef CAS PubMed; (f) X. Y. Mak, P. Laurino and P. H. Seeberger, Beilstein J. Org. Chem., 2009, 5 DOI:10.3762/bjoc.5.19.
- (a) P. Dambruoso, M. Ballestri, C. Ferroni, A. Guerrini, G. Sotgiu, G. Varchi and A. Massi, Green Chem., 2015, 17, 1907–1917 RSC; (b) O. Bortolini, A. Cavazzini, P. P. Giovannini, R. Greco, N. Marchetti, A. Massi and L. Pasti, Chem. – Eur. J., 2013, 19, 7802–7808 CrossRef CAS PubMed; (c) O. Bortolini, A. Cavazzini, P. Dambruoso, P. P. Giovannini, L. Caciolli, A. Massi, S. Pacifico and D. Ragno, Green Chem., 2013, 15, 2981–2992 RSC; (d) O. Bortolini, L. Caciolli, A. Cavazzini, V. Costa, R. Greco, A. Massi and L. Pasti, Green Chem., 2012, 14, 992–1000 RSC; (e) A. Massi, A. Cavazzini, L. Del Zoppo, O. Pandoli, V. Costa, L. Pasti and P. P. Giovannini, Tetrahedron Lett., 2011, 52, 619–622 CrossRef CAS; (f) A. Massi, O. Pandoli, A. Cavazzini, L. Del Zoppo, P. P. Giovannini and C. Bendazzoli, Italian Patent, 0001398243, 2013 Search PubMed.
- (a) G. Jas and A. Kirschning, Chem. – Eur. J., 2003, 9, 5708–5723 CrossRef CAS PubMed; (b) A. Stankiewicz, Chem. Eng. Sci., 2001, 56, 359–361 CrossRef CAS.
- (a) R. M. Heck, S. Gulati and R. J. Farratu, Chem. – Eur. J., 2001, 82, 149–156 CAS; (b) S. Ceylan and A. Kirschning, in Recoverable and Recyclable Catalysts, ed. M. Benaglia, John Wiley and Sons, 2009, ch. 13, pp. 379–403 Search PubMedFor recent reports on monolithic microreactors, see: (c) R. J. Ingham, E. Riva, N. Nibkin, I. R. Baxendale and S. V. Ley, Org. Lett., 2012, 14, 3920–3923 CrossRef CAS PubMedand references therein (d) H. Lange, M. J. Capener, A. X. Jones, C. J. Smith, N. Nibkin, I. R. Baxendale and S. V. Ley, Synlett, 2011, 869–873 CAS; (e) B. Ngamson, A. M. Hickey, G. M. Greenway, J. A. Littlechild, P. Watts and C. Wiles, J. Mol. Catal. B: Enzym., 2010, 63, 81–86 CrossRef.
- S. Martín, R. Porcar, E. Peris, M. Isabel Burguete, E. García-Verdugo and S. V. Luis, Green Chem., 2014, 16, 1639–1647 RSC.
- V. Chiroli, M. Benaglia, A. Puglisi, R. Porta, R. P. Jumdeb and A. Mandoli, Green Chem., 2014, 16, 2798–2806 RSC.
- R. Mahrwald, Modern Methods in Stereoselective Aldol Reactions, Wiley-VCH, Weinheim, 2013 Search PubMed.
- A. L. W. Demuynck, L. Peng, F. de Clippel, J. Vanderleyden, P. A. Jacobs and B. F. Sels, Adv. Synth. Catal., 2011, 353, 725–732 CrossRef CAS.
- C. Ayats, A. H. Henseler and M. A. Pericàs, ChemSusChem, 2012, 5, 320–325 CrossRef CAS PubMed.
- S. B. Ötvös, I. M. Mándity and F. Fülöp, J. Catal., 2012, 295, 179–185 CrossRef.
- G. Rulli, K. A. Fredriksen, N. Duangdee, T. Bonge-Hansen, A. Berkessel and H. Gröger, Synthesis, 2013, 45, 2512–2519 CrossRef CAS.
- A. Gurka, I. Bucsi, L. Kovács, G. Szőllősi and M. Bartók, RSC Adv., 2014, 4, 61611–61618 RSC.
- For a recent study on a 3D continuous-flow organocatalytic system with on-line reaction monitoring, see: G. S. Scatena, A. F. de la Torre, Q. B. Cass, D. G. Rivera and M. W. Paixão, ChemCatChem, 2014, 6, 3208–3214 CrossRef CAS.
- For the use of homogeneous (S)-5-(pyrrolidin-2-yl)-1H-tetrazole in organocatalytic strategies, see: (a) D. A. Longbottom, V. Franckevičius, S. Kumarn, A. J. Oelke, V. Wascholowski and S. V. Ley, Aldrichimica Acta, 2008, 41, 3–11 CAS; (b) S. Kumarn, A. J. Oelke, D. M. Shaw, D. A. Longbottom and S. V. Ley, Org. Biomol. Chem., 2007, 5, 2678–2689 RSC; (c) A. J. A. Cobb, D. M. Shaw, D. A. Longbottom, J. B. Gold and S. V. Ley, Org. Biomol. Chem., 2005, 3, 84–96 RSC; (d) V. Franckevičius, K. R. Knudsen, M. Ladlow, D. A. Longbottom and S. V. Ley, Synlett, 2006, 889–892 Search PubMed; (e) A. J. Oelke, S. Kumarn, D. A. Longbottom and S. V. Ley, Synlett, 2006, 2548–2552 CAS; (f) C. E. T. Mitchell, A. J. A. Cobb and S. V. Ley, Synlett, 2005, 611–614 CAS; (g) A. J. A. Cobb, D. M. Shaw and S. V. Ley, Synlett, 2004, 558–560 CAS; (h) A. Hartikka and P. I. Arvidsson, Tetrahedron: Asymmetry, 2004, 15, 1831–1834 CrossRef CAS; (i) A. Hartikka and P. I. Arvidsson, Eur. J. Org. Chem., 2005, 4287–4295 CrossRef CAS; (j) H. Torii, M. Nakadai, K. Ishihara, S. Saito and H. Yamamoto, Angew. Chem., Int. Ed., 2004, 43, 1983–1986 CrossRef CAS PubMed; (k) N. Momiyama, H. Torii, S. Saito and H. Yamamoto, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5374–5378 CrossRef CAS PubMed; (l) N. S. Chowdari, M. Ahmad, K. Albertshofer, F. Tanaka and C. F. Barbas III, Org. Lett., 2006, 8, 2839–2842 CrossRef CAS PubMed.
- (a) Z. An, Y. Guo, L. Zhao, Z. Li and J. He, ACS Catal., 2014, 4, 2566–2576 CrossRef CAS; (b) M. Gruttadauria, A. M. P. Salvo, F. Giacalone, P. Agrigento and R. Noto, Eur. J. Org. Chem., 2009, 5437–5444 CrossRef CAS.
- E. C. Peters, F. Svec and J. M. Fréchet, Adv. Mater., 1999, 11, 1169–1181 CrossRef CAS.
- R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437–1451 RSC.
- F. E. Valera, M. Quaranta, A. Moran, J. Blacker, A. Armstrong, J. T. Cabral and D. G. Blackmond, Angew. Chem., Int. Ed., 2010, 49, 2478–2485 CrossRef CAS PubMed.
- N. Zotova, A. Franzke, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2007, 129, 15100–15101 CrossRef CAS PubMed.
- Y. Li, H. D. Tolley and M. L. Lee, Anal. Chem., 2009, 81, 9416–9424 CrossRef CAS PubMed.
- The FT-IR spectrum of the monolithic catalyst MM6 after utilization under continuous-flow conditions for 150 h was almost identical to that of the freshly prepared catalyst. It is therefore reasonable to speculate that the observed progressive loss of activity after that time was due to the interference of impurities accumulated onto the monolithic catalyst. Attempts to regenerate the optimal activity by washing the column with TFA and Et3N solutions were unsuccessful.
- V. A. Soloshonok, Angew. Chem., Int. Ed., 2006, 45, 766–769 CrossRef CAS PubMed.
- R. M. McCormick and B. L. Karger, Anal. Chem., 1980, 52, 2249–2257 CrossRef CAS.
- F. Gritti, Y. Kazakevich and G. Guiochon, J. Chromatogr. A, 2007, 1161, 157–169 CrossRef CAS PubMed.
- V. Costa, L. Pasti, N. Marchetti, F. Dondi and A. Cavazzini, J. Chromatogr. A, 2010, 1217, 4919–4924 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 1–4, FT-IR spectra of M5–M6, SEM image of MM6, chiral HPLC chromatograms of 9–11. See DOI: 10.1039/c5re00017c |
| This journal is © The Royal Society of Chemistry 2016 |