
The substitution of the platinum counter electrode in a plasmonic photoelectrochemical system with near-infrared absorption for solar water splitting
Abstract
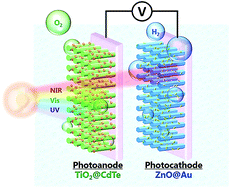
Converting solar energy into a usable chemical fuel has attained great importance in the past decade. The near-infrared and infrared regions contain nearly half of the photons present in the total flux of solar irradiation; however, near-infrared-active and infrared-active materials cannot commonly provide sufficient potential for electron–hole pairs to drive the photoelectrochemical reaction, which limits the development of efficient solar energy conversion devices for future applications. Here, we report on a photoelectrochemical cell that is constructed using ZnO@Au rod nanostructures as the photocathode and TiO2@CdTe quantum dot nanostructures as the photoanode. In this cell, the photoactive materials can utilize a wide range of the solar spectrum (up to the near-infrared). Using a plasmonic photocathode, a maximum efficiency of about 1.4% (at +0.5 V) was exhibited, which was comparable to that attained when using conventional Pt foil as the electrodes. The use of plasmonic materials has several advantages, including easily customizable optical properties and the ability for coupling with plasmon-inducing electromagnetic fields/hot electrons, which can effectively enhance the photocatalytic water splitting reactions. This research aims to provide an alternative photoelectrode to replace conventional Pt electrodes, to improve the conversion efficiency for solar energy.
 Please wait while we load your content...
Please wait while we load your content...

