
Silicon oxycarbide ceramics as anodes for lithium ion batteries: influence of carbon content on lithium storage capacity†
Abstract
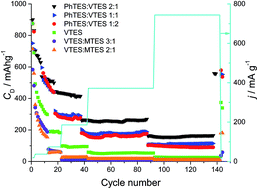
We report here on the synthesis and characterization of silicon oxycarbide (SiOC) in view of its application as a potential anode material for Li-ion batteries. SiOC ceramics are obtained by pyrolysis of various polysiloxanes synthesized by sol–gel methods. The polysiloxanes contain different organic groups attached to silicon, which influence the chemical composition and the microstructure of the final ceramic product. The structure of the SiOC samples is investigated by XRD, micro-Raman spectroscopy, solid state 29Si MAS-NMR and TEM. All investigated samples remain amorphous. However, at the elevated temperature of pyrolysis a phase separation process begins. During this process the carbon clusters become more ordered, which is reflected in the higher intensity and narrowing of the D1 band and decreasing of the D3 band. Moreover, the elevated temperature of pyrolysis promotes consumption of mixed bonds units, SiO3C, SiO2C2, SiOC3, and increases the share of oxygen rich SiO4 and carbon rich SiC4 tetrahedra. Electrochemical studies show a clear dependence between free carbon content and lithium storage capacity. Carbon-rich samples exhibit significantly higher capacities (∼550 mA h g−1 recorded at low current rate after 140 charge–discharge cycles) compared to carbon-poor samples (up to 360 mA h g−1). Moreover, carbon-rich samples exhibit a lower irreversible capacity during their first cycles compared to low carbon samples.
 Please wait while we load your content...
Please wait while we load your content...

