
Evaluation of graphene-wrapped LiFePO4 as novel cathode materials for Li-ion batteries†
Abstract
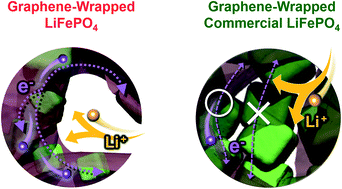
Graphene-wrapped LiFePO4 (LiFePO4/G) is introduced as a cathode material for Li-ion batteries with an excellent rate capability. A straightforward solid-state reaction between graphene oxide-wrapped FePO4 and a lithium precursor resulted in highly conducting LiFePO4/G composites, which feature ∼70 nm-sized LiFePO4 crystallites with robust connection to the external graphene network. This unique morphology enables all LiFePO4 particles to be readily accessed by electrons during battery operation, leading to a remarkably enhanced rate capability. The in situ electrochemical impedance spectra were studied in detail throughout charge and discharge processes, by which enhanced electronic conductance and thereby reduced charge transfer resistance was confirmed as the origin of the superior performance in the novel LiFePO4/G.
 Please wait while we load your content...
Please wait while we load your content...

