
Discovery of novel cinnamylidene-thiazolidinedione derivatives as PTP-1B inhibitors for the management of type 2 diabetes†‡
Abstract
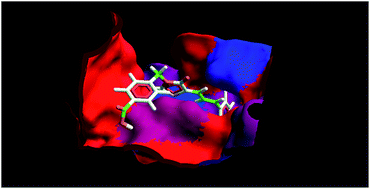
Protein tyrosine phosphatase-1B (PTP-1B) inhibition is a legitimate approach to combat type 2 diabetes and obesity as it corrects the insulin and leptin signalling cascades. In pursuing this, our goal is to discover compounds bearing small molecular scaffolds, and able to inhibit PTP-1B in a selective manner. In the present work, we have synthesized N3-substituted cinnamylidene-thiazolidinediones (4p–4x), and evaluated in vitro PTP-1B inhibitory activity, and in vivo anti-hyperglycaemic potential in streptozotocin-nicotinamide induced diabetic mice. Among various synthesized compounds, 4w exhibited the most potent in vitro PTP-1B inhibitory activity (IC50 ∼ 6.52 μM) along with excellent in vivo anti-hyperglycaemic activity. Furthermore, molecular docking assisted 3D-QSAR study was performed on the synthesized compounds (4p–4x) for the exploration of the binding mode of interactions, prediction of the binding affinity, and identification of 3D-pharmacophoric features (steric and electrostatic) responsible for inhibitory activity. The docking results were in agreement with the biological activity results i.e. compound 4w showed the highest binding affinity with PTP-1B (MolDock score −123.715), which is comparable with ertiprotafib (MolDock score −125.183). The lead discovered can be used for the further development of cinnamylidene-thiazolidinedione derivatives as antidiabetic agents.
 Please wait while we load your content...
Please wait while we load your content...

