
Stereoelectronic effects of the glycosidic linkage on the conformational preference of d-sucrose†
Abstract
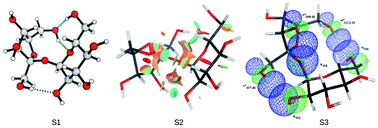
The stereoelectronic effects involved in the conformations shown by D-sucrose were analysed at the M06-2X/6-31++G(d,p) level, both under vacuum and in water with the continuum solvation model IEF-PCM, and employing the natural bond orbital theory (NBO) and non-covalent interactions (NCI). Different groups of conformations for D-sucrose in relation to the glycosidic linkage were evaluated and the results showed that not only were hydrogen bonds important to explain the relative energy observed, but it was also necessary to consider any stabilizing orbital interactions involving the glycosidic linkage. The most stable conformation observed in water had dihedral angles for the glycosidic linkage with values of 110.8° and −44.9° for ϕ and ψ, respectively.
 Please wait while we load your content...
Please wait while we load your content...

