
Remarkable enhancement of the catalytic performance of molybdenum sulfide catalysts via an in situ decomposition method for higher alcohol synthesis from syngas†
Na Wangabc,
Jianli Lib,
Xuemin Liub,
Ruijue Hub,
Yulong Zhangb,
Haiquan Su*ab and
Xiaojun Gu*b
aChemical Engineering College, Inner Mongolia University of Technology, Hohhot 010051, Inner Mongolia, China. E-mail: haiquansu@yahoo.com
bInner Mongolia Key Laboratory of Coal Chemistry, School of Chemistry and Chemical Engineering, Inner Mongolia University, Hohhot 010021, Inner Mongolia, China. E-mail: xiaojun.gu@yahoo.com
cBaotou Teachers'College, Baotou 014030, Inner Mongolia, China
First published on 21st November 2016
Abstract
Nickel-containing and nickel-free molybdenum sulfide catalysts were prepared by an in situ thermal decomposition method. Their catalytic performance was investigated for higher alcohol synthesis from syngas. It is surprising that the Ni-promoted and the nickel-free catalysts prepared by in situ decomposition showed significantly higher activity featuring space-time-yield (STY, mg g−1 h−1) values of 520 and 510 respectively, while the ex situ method resulted in K–Ni–MoS2-E and K–MoS2-E catalysts with low activities featuring STY values of 370 and 170 mg g−1 h−1, respectively. The remarkable difference in catalytic activities was due to the fact that the in situ decomposition method can result in disordered MoS2 domains and exposure to more coordinated unsaturated sites by the formation of smaller particles. In addition, the excellent catalytic performance was also associated with the combined impact of the in situ preparation method and Ni promotion.
1. Introduction
The catalytic conversion of syngas (CO and H2) to higher alcohols has attracted worldwide attention as a renewable and environmentally-friendly fuel for reducing global warming and improving global energy security.1–3 Moreover, higher alcohol is an alternative fuel to improve the octane number of gasoline,4–6 and also has the advantages of lower vapor pressure, better miscibility with hydrocarbon compounds, improved water tolerance, higher overall heating value and less emission of pollutants.7–9 Nevertheless, the existing technology of higher alcohols synthesis is still on a small scale. The single-pass-conversion of the feed syngas and selectivity to total alcohols and C2+OH are both relatively low. The application of such an attractive synthesis still suffers from the lack of high performance catalysts. As a result, several catalytic systems have been investigated for higher alcohols synthesis.10–15 Among various catalysts, alkali-modified molybdenum sulfide (ADM) catalysts are more attractive because of their higher selectivity to total alcohols, higher activityfor water–gas shift reaction and excellent resistance to sulfur poisoning for feed gas.16–18 However, their space-time-yield (STY) and selectivity for higher alcohols are usually low, and methanol is the dominant product.19,20 The addition of Ni as a promoter to K-modified MoS2 catalysts is found to be able to greatly enhance the selectivity to C2+ alcohols and the STY of total alcohols,21–23 which are related to the dispersion improvement, structure modification of active phase and the strong ability of CO insertion.20,23–26Although the detailed mechanism of higher alcohols synthesis from syngas is still unclear so far, many research results have indicated that the catalytic activity of alkali-modified MoS2 catalysts is strongly dependent on the ratio of edge sites to basal plane area of the MoS2 domains, because the coordinated unsaturated sites that may act as active sites on which chemisorption of probe molecules can occur on the edge sites not on the basal planes.27–29 Furthermore, the ratio of edge sites to basal plane area of the MoS2 domains is proportional to the exfoliated degree of stacking layers of MoS2. In general, ADM catalysts are normally prepared through ex situ method with precipitation or impregnation and high temperature calcination procedure. It is easy to form bulk MoS2 that leads to the decrease ratio of edge sites to basal plane area and is unfavorable for the higher alcohols synthesis.30,31 Thus, exploring an appropriate method synthesis of sulfide molybdenum-based catalysts to increase the ratio of edge sites and combining it with the promotion effect of promoters may be an effective strategy for improving the catalytic performance of molybdenum sulfide-based catalyst.
Here we reported an efficient catalytic system resulted from the combined impact of promoters and preparation method for higher alcohols synthesis. Surprisingly, catalyst containing Ni-promoters prepared by in situ decomposition method produced huge amount of smaller MoS2 slabs and significantly increased the exfoliation of MoS2 layers, leading to exposing large amounts of coordinated unsaturated sites. As a result, the catalytic activity remarkably increased, and the highest higher alcohols selectivity (58.72%) and STY of total alcohols (520 mg g−1 h−1) were obtained on the K–Ni–MoS2–I catalyst.
2. Experimental section
2.1 Chemicals
All chemicals were obtained from commercial sources and used without further purification. Ammonium heptamolybdate ((NH4)2Mo7O24·4H2O, Sinopharm Chemical Reagent Co., Ltd, >99%), Ammonium sulphide ((NH4)2S, Alfa Aesar, 20% aq. soln.), potassium carbonate (K2CO3, Alfa Aesar, >99%), nickel(II) acetate tetrahydrate (Ni(CH3COO)2·4H2O, Sinopharm Chemical Reagent Co., Ltd, >99%).2.2 Catalyst preparation
(NH4)2MoS4 was obtained through the reaction of (NH4)2S, (NH4)2Mo7O24·4H2O.32 A weight of 4 g of ammonium heptamolybdate (NH4)2Mo7O24·4H2O is dissolved in 20 mL of ammonium hydroxide. To this solution is added 38.4 mL of concentrated ammonium sulfide. The mixture is heated at 333 K for 1 h then cooled in an ice bath for 3 h to induce crystallization. Crystals are separated by filtration, washed with isopropyl alcohol and left to dry at room temperature.The nickel-containing and nickel-free molybdenum sulfide catalysts were prepared by in situ thermal decomposition method, which were labeled as K–MoS2–I and K–Ni–MoS2–I. The molar ratio of K to Mo in the two catalysts was 0.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. K–MoS2–I catalyst was prepared by grinding (NH4)2MoS4 with K2CO3 in a mortar and pestling in ether for 1 h, the resulting mixture was dried. Before reaction, catalysts were decomposed in situ by syngas at 10 MPa pressure with WHSV of 3000 h−1 and 330 °C for 12 h. The preparation procedure of K–Ni–MoS2–I was the same as the K–MoS2–I except introduction of Ni(CH3COO)2·4H2O (Ni/Mo molar ratio = 1:3).
:1. K–MoS2–I catalyst was prepared by grinding (NH4)2MoS4 with K2CO3 in a mortar and pestling in ether for 1 h, the resulting mixture was dried. Before reaction, catalysts were decomposed in situ by syngas at 10 MPa pressure with WHSV of 3000 h−1 and 330 °C for 12 h. The preparation procedure of K–Ni–MoS2–I was the same as the K–MoS2–I except introduction of Ni(CH3COO)2·4H2O (Ni/Mo molar ratio = 1:3).
For comparison, K–MoS2-E and K–Ni–MoS2-E catalysts (K/Mo molar ratio = 0.7:1) were prepared by ex situ method. A typical procedure was as follows: firstly, MoS2 was obtained by thermal decomposition of the (NH4)2MoS4 at a temperature rate of 5 °C min−1 to 500 °C and holding for 1 h under argon atmosphere. Ni–MoS2 catalyst precursor was formed by mechanically mixed (NH4)2MoS4 with Ni(CH3COO)2·4H2O (Ni/Mo molar ratio = 1:3) in a mortar and pestle in ether for 1 h, the resulting mixture was dried. The decomposition procedure of Ni–MoS2 catalyst precursor was the same as MoS2. Then, K–MoS2-E and K–Ni–MoS2-E catalysts were prepared by grounding K2CO3 together with MoS2 or Ni–MoS2 catalyst precursor in a mortar and pestle, respectively. In addition, in order to eliminate the influence factors of calcination temperature of catalysts by ex situ method, K–MoS2-E(330) and K–Ni–MoS2-E(330) were synthesized using the similar process to that of K–MoS2-E except calcination temperature at 330 °C (see the ESI†).
2.3 Characterization
Powder X-ray diffraction (PXRD) studies were conducted using a Panalytical X-Pert X-ray diffractometer with a Cu-Kα source (40 kV, 20 mA). The transmission electron microscopy (TEM, FEI Tecnai G2 F20) equipped with an energy dispersive X-ray (EDX) spectroscopy was applied for the morphologies and composition of the as-synthesized samples. Temperature programmed reduction (TPR) experiments were carried out in a Micromeritics AutoChem II 2920 automatic analyzer equipped with a TCD and the reactor effluent composition was continuously monitored as a function of sample temperature by a mass spectrometer (Hiden, QIC-20). Prior to the experiments, catalysts (0.08 g) were activated for 1 h in Ar at 330 °C, and then cooled to room temperature under the same gas flow. The reduction step was performed in 10% H2 in Ar, with a heating rate of 5 °C min−1 up to 900 °C. Detailed analyses for catalytic products were performed on gas chromatography (GC) (Shimadzu GC-2014).2.4 Catalytic performance tests
The catalytic test for higher alcohol synthesis was conducted at 10.0 MPa, 330 °C, 3000 h−1 and H2 to CO molar ratio of 1 in a fixed-bed stainless-steel reactor. For each experiment, 2 g of the catalyst diluted with 20 mL of 40–60 mesh size silicon sand was loaded and activated in flowing syngas at 330 °C for 12 h before the data acquisition. The gaseous and liquid products were on-line analyzed on GC. The mixed alcohols were calculated directly from the peak areas by using a standard liquid of C1–C5 mixed alcohols.Mass balance calculations were used to calculate the % CO conversion, product STY, and alcohol selectivity (mol%). The conversion of CO (XCO) is calculated from the molar flow rates of CO (FCO) into and out of the reactor:
 | (1) |
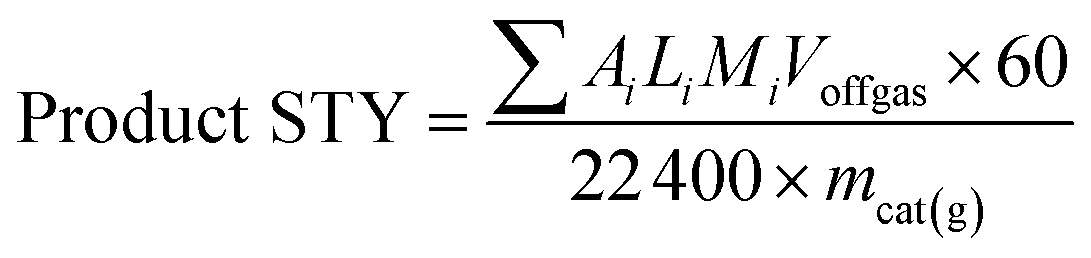 | (2) |
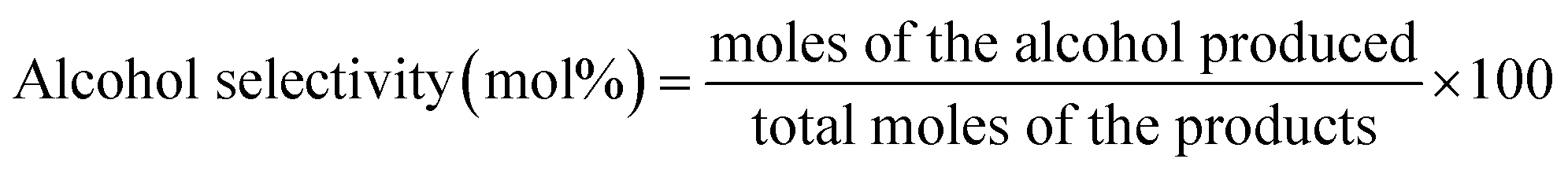 | (3) |
3. Results and discussion
3.1 PXRD analysis
The PXRD patterns of the samples after reaction showed the typical peaks of MoS2 crystallites in Fig. 1. The diffraction peak at 2θ ≈ 14° corresponds to the (0 0 2) basal planes of MoS2.33–35 An interesting difference between the catalysts can be observed for the intensity of the (0 0 2) planes of MoS2 domains. Clearly, the characteristic diffraction peak of the (0 0 2) planes of MoS2 domains almost disappeared on the catalysts prepared by in situ decomposition method compared to the catalysts prepared by ex situ method, illuminating that it forms a well-dispersed MoS2 crystallized structure and a much smaller size of (002) basal planes of MoS2,33,35–37 which was confirmed by the TEM analysis.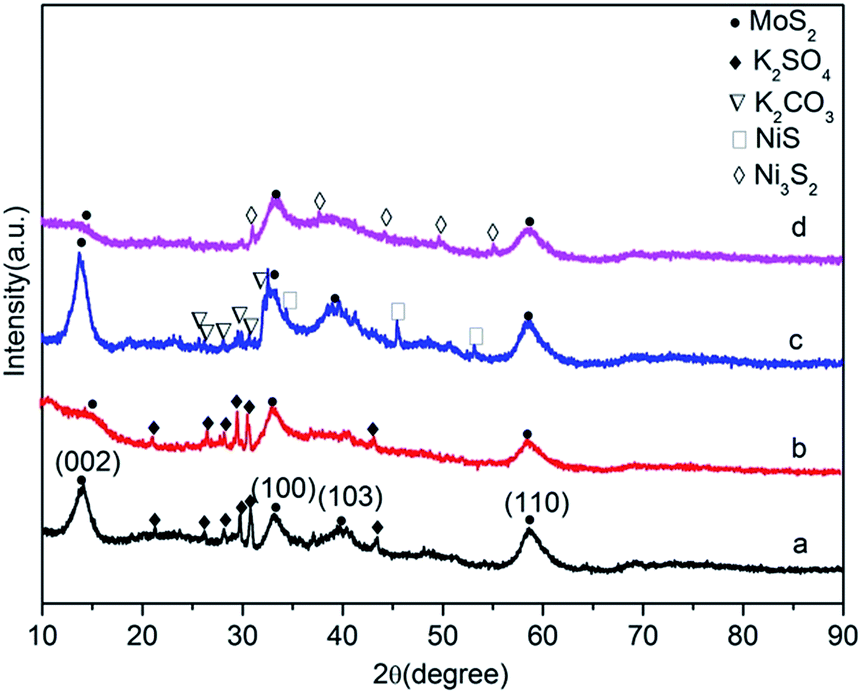 | ||
| Fig. 1 PXRD patterns of the spent catalysts: (a) K–MoS2-E; (b) K–MoS2–I; (c) K–Ni–MoS2-E; (d) K–Ni–MoS2–I. | ||
Obviously, the XRD patterns of K–MoS2–I and K–Ni–MoS2–I catalysts were composed of two broad peaks, corresponding to (1 0 3) and (1 1 0) diffractions, respectively, indicating that in situ method resulted in poorly crystalline MoS2 particles. In addition, Bragg scattering from the layer-disordered crystallites has been investigated theoretically,38,39 and the diffuse natures of the (h 0 l) peaks are mainly considered to be related to rotational or turbostratic disorder of layers in MoS2 crystals.40 As is shown in Fig. 1, the absent of (1 0 3) peak of K–MoS2–I and K–Ni–MoS2–I catalysts demonstrates that individual MoS2 layers are rotated and shifted against each other (turbostratic disorder), indicating that the in situ decomposition was favorable for the formation of disordered structure of MoS2 slabs, which can increase the active sites of the catalyst.41,42
In order to better understand the difference in preparation methods, the preparation procedure of the catalysts and their dominant phases after reaction characterized by PXRD are summarized in Table 1. In K–MoS2-E and K–MoS2–I catalysts, K2SO4 species were observed at 26.2°, 28.1°, 29.6°, and 30.7° (PDF number: 05-0613), which indicates that K phase was transformed into K2SO4 phase by exposing air after reaction.43 (Detailed discussion in the TPR sections of ESI†). For K–Ni–MoS2-E catalyst prepared by ex situ method, the addition of Ni caused diffraction peaks at 2θ values of 34.4°, 45.5° and 53.2°, which were related to the separated phase of NiS. However, the weak diffraction peak of Ni3S2 was observed in K–Ni–MoS2–I compared with that of K–Ni–MoS2-E, indicating that in situ decomposition method effectively inhibited the formation of separated nickel sulfides.24 This was in favor of forming alcohols, because separated nickel sulfides were active for the formation of hydrocarbons.24 Moreover, the intact K2CO3 phase was only found in the pattern of K–Ni–MoS2-E. It could be ascribed to poorly dispersed MoS2 domains leading to lower exposure of active sites, which were verified by the PXRD analyses with the observation of the highest intensity of the (0 0 2) planes in all catalysts.
| Catalyst | Crystal phase | 2θ |
|---|---|---|
| K–MoS2-E | MoS2 | 14.1, 33.1, 39.9, 58.7 |
| K2SO4 | 26.2, 28.1, 29.6, 30.7, 43.4 | |
| K–MoS2–I | MoS2 | 33.1, 58.6 |
| K2SO4 | 26.2, 28.1, 29.6, 30.7 | |
| K–Ni–MoS2-E | MoS2 | 13.7, 33.0, 39.4, 58.6 |
| NiS | 34.4, 45.5, 53.2 | |
| K2CO3 | 26.3, 28.0, 29.6, 30.7, 32.1 | |
| K–Ni–MoS2–I | MoS2 | 33.2, 58.6 |
| Ni3S2 | 31.1, 37.6, 44.2, 49.7, 55.0 |
3.2 TEM analysis
Transmission electron microscopy (TEM) analysis revealed the morphology and structure changes of MoS2 domains for in situ decomposition method and ex situ method. In K–MoS2-E (Fig. 2a), MoS2 crystallites exhibited very uniform patterns with long and straight multi-layered structure. Once in situ decomposition method was used (Fig. 2b), the K–MoS2–I catalyst showed an estimated slab length of about 6–9 nm, which is much shorter than that of K–MoS2-E (30–100 nm). Thus, the amount of the MoS2 slabs in K–MoS2–I catalyst is far more than that in K–MoS2-E. In addition, the average number of stacked MoS2 layers along the c-axis in K–MoS2–I (5 to 6 stacked layers) is lower than that in K–MoS2-E (10 to 25 stacked layers). Moreover, the MoS2 slabs in K–MoS2–I are more disordered than that in K–MoS2-E. All these results are in good agreement with the PXRD analyses and suggest that in situ decomposition method was favorable for the formation of smaller particles leading to the exposure of more edge and corner sites over K–MoS2–I.44,45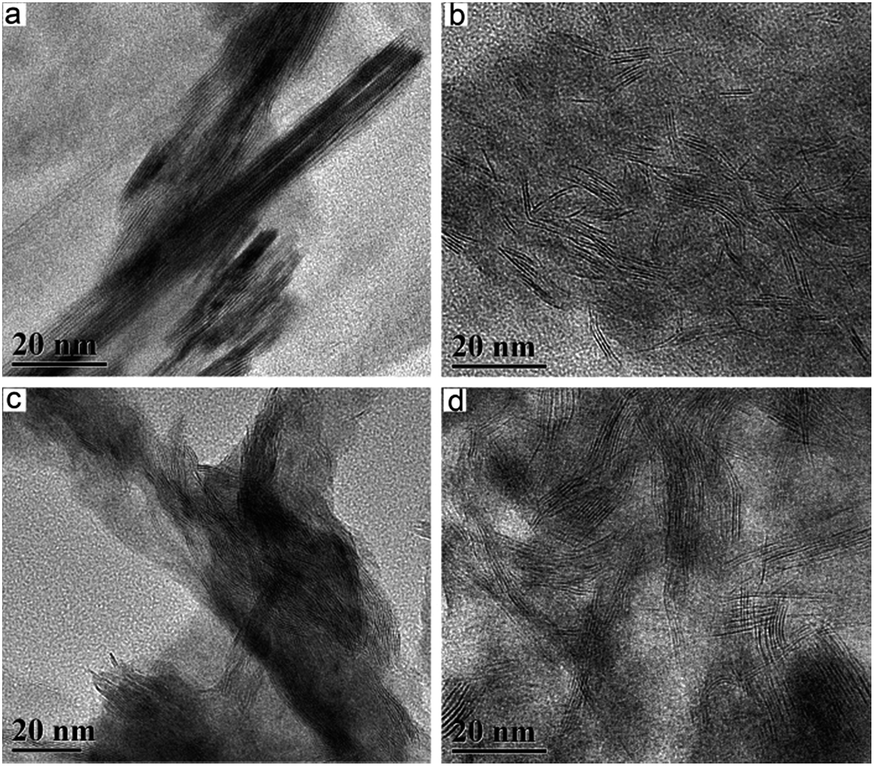 | ||
| Fig. 2 TEM images of the catalysts after reaction: (a) K–MoS2-E; (b) K–MoS2–I; (c) K–Ni–MoS2-E; (d) K–Ni–MoS2–I. | ||
After the addition of Ni, these slabs of MoS2 in K–Ni–MoS2-E became more curved and more stacked than that in K–MoS2–I, indicating that Ni modified the MoS2 structure. Furthermore, these smaller slabs stuck together in bigger agglomerates (Fig. 2c), leading to decreasing the exposure of active sites. It is unfavorable for the formation of alcohols. However, this negative effect could be compensated and improved once the in situ decomposition method was applied. When K–Ni–MoS2–I catalyst was prepared using in situ decomposition method (Fig. 2d), it can be clearly seen that the MoS2 domains was highly dispersed and disorganized and the slab length of MoS2 became shorter. This led to increasing the ratio of edge sites to basal plane area of the MoS2 domains and higher exposure of active sites on the surface of catalysts, which responsible for its better activity because the catalytic reactions are mainly performed on the edges of the slabs.44,45
The schematic structure of MoS2 domains for different catalysts revealed by TEM was shown in Fig. 3. MoS2 domains of these catalysts prepared using in situ decomposition are notably different from those observed in MoS2 crystallites obtained by ex situ method. In situ decomposition resulted in the catalyst with relatively more disordered layers, shorter MoS2 slabs, less stacked layers and better dispersion than ex situ method. All these factors under the in situ condition led to the increase ratio of edge sites to basal plane area of the MoS2 domains and higher exposure of active sites on the surface of catalysts, which were responsible for its better activity.20,46 Additionally, incorporation of Ni into the Mo sulfide catalysts causes a high degree of bending of the MoS2 layers, which may also correspond to an improved activity. The separated NiS phase with the lattice space values of 0.4541 nm of K–Ni–MoS2-E were estimated by the high-resolution TEM (HRTEM) as shown in Fig. 4D), and by a Fast Fourier Transform (FFT) pattern (inset in Fig. 4D). However, separated Ni3S2 phase was not observed, and then the high-angle annular dark-field scanning transmission electron microscopy (HADDF-STEM) and EDX elemental line scan images of K–Ni–MoS2–I (Fig. 4A and B) show that the metallic elements Mo, Ni, S and K are homogeneously dispersed on the surface of K–Ni–MoS2–I catalyst.
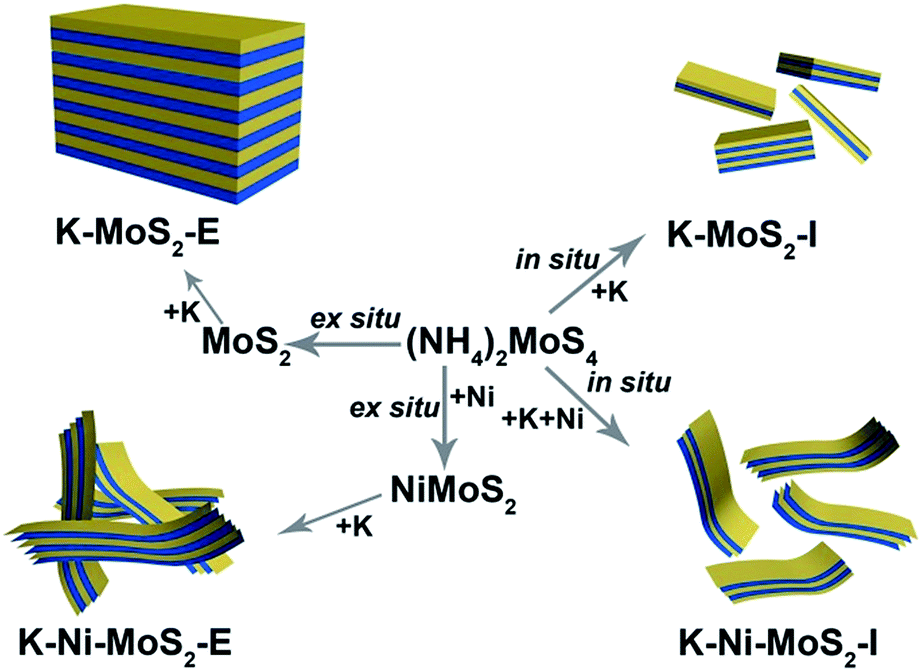 | ||
| Fig. 3 Schematic structures of MoS2 domains for different preparation method of catalysts. | ||
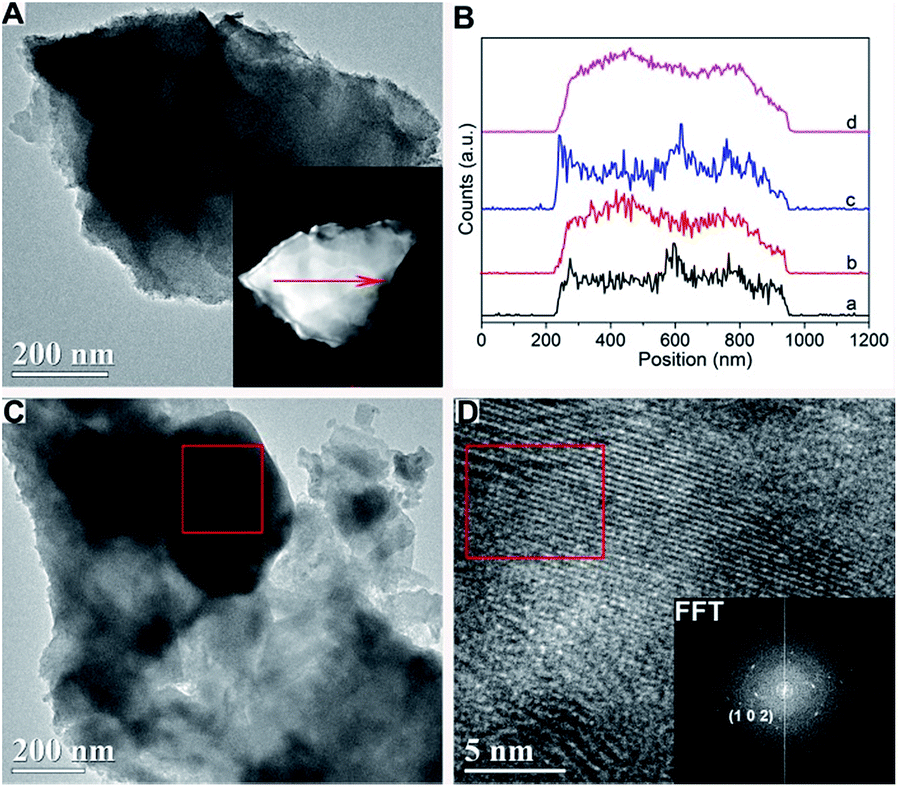 | ||
| Fig. 4 TEM images of the catalysts after reaction: (A) TEM and HAADF-STEM (inset) images of K–Ni–MoS2–I and (B) EDS line scan showing elemental distributions along the arrow marked in the inset of (A): (a)–(d) EDS line spectrum of K–K, Mo–K, Ni–K and S–K, respectively. (C) TEM image of K–Ni–MoS2-E. (D) HRTEM image of K–Ni–MoS2-E (zoomed from the red square in Fig. 4C, inset: Fast Fourier Transform (FFT) demonstrating the (102) plane of NiS). | ||
3.3 TPR study
To examine the chemical reactivity of active sites located at the MoS2 layer edges by the removal of sulfur-containing phases released in the form of H2S on the surface of catalysts, the TPR and the corresponding mass signal of the catalysts after in situ activated at 330 °C were measured and shown in Fig. 5. For K–MoS2-E catalyst, the small H2 reduction peak appeared between 127 and 209 °C was associated with the small H2S production peak. Both the reduction and corresponding H2S peaks in this low temperature range could be assigned to the nonstoichiometric sulfur species (Sx) of active phases adsorbed on coordinated unsaturated edge/corner sites of MoS2.47–50 Simultaneously, a weak and flat reduction peak and H2S production peak were observed respectively around 380 °C to 600 °C due to the reduction of more strongly bound S2− ions (stoichiometric sulfides).51 However, once the in situ decomposition method was used, the most significant changes were that the peak corresponding to hydrogenation of Sx disappeared in the same temperature for K–MoS2–I compared to K–MoS2-E (Fig. 5b), while three strong new reduction peaks coincided with the H2S production peaks at the low temperature between 200 °C and 400 °C were observed and increased area under the TPR curves, indicating the amount of Sx increased for K–MoS2–I. Actually, the amount of Sx formed depends on the number of coordinated unsaturated sites, which is related to catalytic activity.52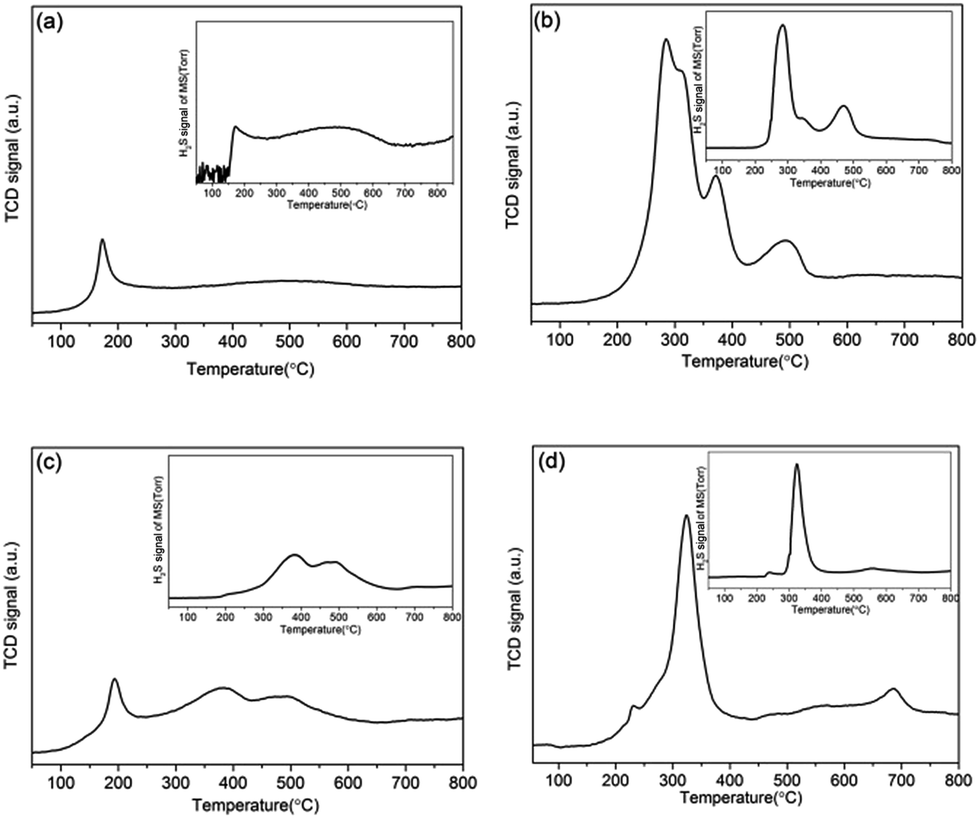 | ||
| Fig. 5 TPR profiles of the catalysts: (a) K–MoS2-E; (b) K–MoS2–I; (c) K–Ni–MoS2-E; (d) K–Ni–MoS2–I after in situ activation at 330 °C. Insets are the corresponding H2S signal of MS. | ||
When Ni promoter was added, K–Ni–MoS2-E catalyst (Fig. 5c) exhibits two peaks before 400 °C. However, the small H2 consumption peak was not accompanied by H2S production peak in the same temperature region, indicating that Ni promoter was sufficiently fast to prevent formation of Sx by the formation of new complex phases, so-called Ni–Mo–S active phase.53 When the in situ decomposition method was applied to K–Ni–MoS2–I catalyst, the strong reduction peak coincided with the H2S production peak at about 200–400 °C appeared, indicating the amount of coordinated unsaturated sites were formed.
Comparing the quantification of the amount of hydrogen consumed in the low temperature region during the reduction of the catalysts (Table 3), we found that the reduction of the samples prepared with in situ method proceeds with higher consumption of hydrogen than of the reference K–MoS2-E and K–Ni–MoS2-E catalysts formed by ex situ method. This is an indication of different amounts of weakly bonded sulfur species existed on the catalyst surface, implying that the samples treated on in situ condition have a larger number of coordinated unsaturated sites, which were considered to be the active sites for the higher alcohol synthesis.54 It was well consistent with PXRD and TEM results.
| Catalyst | Alcohol distribution (mol%) | Hydrocarbon distribution | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MeOH | EtOH | PrOH | BuOH | Other oxya | CH4 | C2H6 | C3H8 | C4H10 | Other hydb | |
| a Other oxygenates: 1-propanol, 1-butanol, 2-methyl-1-propanol, i-butanol.b Other hydrocarbons: ethylene, propylene, butylene. | ||||||||||
| K–MoS2-E | 62.6 | 24.7 | 9.8 | 1.4 | 1.5 | 64.9 | 18.6 | 4.4 | 1.4 | 10.7 |
| K–MoS2–I | 40.7 | 27.9 | 19.9 | 3.2 | 8.3 | 71.4 | 9.1 | 1.9 | 1.0 | 16.6 |
| K–Ni–MoS2-E | 32.8 | 45.8 | 14.8 | 1.9 | 4.7 | 77.7 | 8.4 | 1.7 | 0.5 | 11.7 |
| K–Ni–MoS2–I | 23.3 | 49.2 | 19.9 | 2.9 | 4.7 | 78.5 | 8.1 | 1.7 | 0.5 | 11.2 |
| Catalyst | Temperature at maximum (°C) | H2 consumption (cm3 H2 g−1) |
|---|---|---|
| K–MoS2-E | 172 | 5.53 |
| K–MoS2–I | 284 | 56.5 |
| K–Ni–MoS2-E | 194 | 5.8 |
| K–Ni–MoS2–I | 321 | 53.0 |
3.4 Catalytic performance
The catalytic performance of MoS2, K–MoS2-E, K–MoS2–I, K–Ni–MoS2-E and K–Ni–MoS2–I was shown in Fig. 6. The detailed reaction results are summarized in Tables 2 and 4. As seen from the Table 4, MoS2 itself produce only hydrocarbons (primarily methane). However, with the addition of K, the selectivity shifts from hydrocarbon to alcohol. The function of K is to decrease the surface acidity of the catalyst, thus suppressing various side reactions, as well as increasing nondissociative adsorption of CO and reducing the ability to activate hydrogen on the MoS2 surface through site blocking on the sulfide surface.45,55,56 Moreover, K promoter under syngas environment disperses itself uniformly on the MoS2 surface, forming active species for the alcohol synthesis.57 As shown in Tables 2 and 4, it can be seen that the distribution of alcohols over these catalysts is mainly a mixture of C1–C4 linear alcohol, and methane is the main product in the hydrocarbon products for all the catalysts.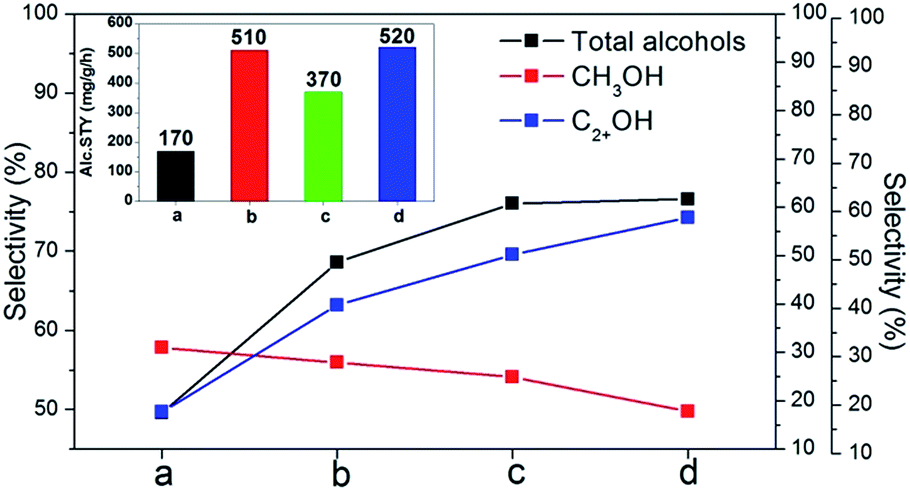 | ||
| Fig. 6 Catalytic performance of the catalysts for higher alcohols synthesis: (a) K–MoS2-E; (b) K–MoS2–I; (c) K–Ni–MoS2-E; (d) K–Ni–MoS2–I. Inset is the corresponding the STY of total alcohols. | ||
For K–MoS2–I, the CO conversion, the selectivity of total alcohols and higher alcohols selectivity and the STY of total alcohols increased respectively by 26%, 38%, 119% and 200% comparable to K–MoS2-E catalyst. This remarkable catalytic activity enhancement could be attributed to the highly dispersed and disordered structure of MoS2 slabs and more MoS2 slabs with coordinated unsaturated sites in K–MoS2–I, because these parameters were closely associated with the active sites for the higher alcohols synthesis.
Over the K–MoS2-E catalyst, the STY of the total alcohols and selectivity to alcohols were rather low, and the major products were hydrocarbons (Table 4). However, when Ni was added to K–MoS2-E catalyst, the catalytic activity markedly increased, which could be due to the Ni promotion effect arisen from the formation of new active phases and bend structure of MoS2 slabs. Once both in situ decomposition method and Ni promotion effect were combined, the catalytic activity was further improved. The CO conversion, the selectivity of total alcohols and higher alcohols selectivity and the STY of total alcohols for K–Ni–MoS2–I catalyst increased respectively by 25%, 55%, 216% and 206% comparable to K–MoS2-E catalyst, which was due to the combined impact of in situ decomposition method and Ni promotion effect in K–Ni–MoS2–I.
4. Conclusions
In summary, the in situ decomposition method has been proven to have many advantages when applying to the preparation of K–MoS2 catalyst. First, it is favorable for producing huge amount of smaller MoS2 slabs, which significantly increases the ratio of edge sites to basal plane area, leading to the exposure of more edges and corners of MoS2, thus enhancing the number of coordinated unsaturated sites, and therefore increasing active sites for alcohols formation. Second, it increases the exfoliation of MoS2 layers and makes the MoS2 slabs more disordered, which plays the same role as the first point. On the other hand, although the addition of Ni as a promoter to K-modified MoS2 catalysts using ex situ method is unfavorable for the exposure of more active sites due to particles conglomerating together, it is able to help form new active phases, which can greatly enhance the selectivity to C2+ alcohols and total alcohols. Third, once the advantages of in situ preparation method and Ni promotion effect were combined, the combined impact between them drastically enhances the catalytic activity. The CO conversion, the selectivity of total alcohols and higher alcohols selectivity and the STY of total alcohols for K–Ni–MoS2–I increased respectively by 25%, 55%, 216% and 206% comparable to K–MoS2-E. Hence, this work provides a promise for designing highly efficient catalyst for higher alcohols synthesis from syngas.Acknowledgements
We are grateful to the Natural Science Foundation of China (21476118), Program for Science and Technology Leading Talent and Innovation Team of Inner Mongolia (20120101) and Prairie Excellence Innovation and Entrepreneurial Team of Inner Mongolia (201201).Notes and references
- G. Liu, D. Pan, T. Niu, A. Cao, Y. Yue and Y. Liu, RSC Adv., 2015, 5, 31637–31647 RSC.
- V. R. Surisetty, A. K. Dalaia and J. Kozinski, Appl. Catal., A, 2011, 404, 1–11 CAS.
- Y. Liu, C. Liu, X. Deng and W. Huang, RSC Adv., 2015, 5, 99023–99027 RSC.
- C. Sun, D. Mao, L. Han and J. Yu, RSC Adv., 2016, 6, 55233–55239 RSC.
- K. Simeonov, J. H. Kim, D. Ferrari, D. Huelsman, G. Budroni, S. Corthals and Y. J. Pagan-Torres, Catal. Sci. Technol., 2014, 4, 922–924 CAS.
- J. Wang, X. Zhang, Q. Sun, S. Chan and H. Su, Catal. Commun., 2015, 61, 57–61 CrossRef CAS.
- S. Velu, N. Satoh, C. S. Gopinath and K. Suzuki, Catal. Lett., 2002, 82, 145–152 CrossRef CAS.
- R. Andersson, M. Boutonnet and S. Järås, Appl. Catal., A, 2012, 417–418, 119–128 CrossRef CAS.
- R. Andersson, M. Boutonnet and S. Järås, Fuel, 2013, 107, 715–723 CrossRef CAS.
- P. Forzatti, E. Tronconi and I. Pasquon, Catal. Rev.: Sci. Eng., 1991, 33, 109–168 CAS.
- I. Wender, Fuel Process. Technol., 1996, 48, 189–297 CrossRef CAS.
- K. Fang, D. Li, M. Lin, M. Xiang, W. Wei and Y. Sun, Catal. Today, 2009, 147, 133–138 CrossRef CAS.
- M. Ojeda, M. L. Granados, S. Rojas, P. Terreros, F. J. Garcia-Garcia and J. L. G. Fierro, Appl. Catal., A, 2004, 261, 47–55 CrossRef CAS.
- T. Jiang, Y. Q. Niu and B. Zhong, Fuel Process. Technol., 2001, 73, 175–183 CrossRef CAS.
- K. Xiao, Z. Bao, X. Qi, X. Wang, L. Zhong, M. Lin, K. Fang and Y. Sun, Catal. Commun., 2013, 40, 154–157 CrossRef CAS.
- W. Xie, J. Zhou, L. Ji, S. Sun, H. Pan, J. Zhu, C. Gao and J. Bao, RSC Adv., 2016, 6, 38741–38745 RSC.
- V. R. Surisetty, I. Eswaramoorthi and A. K. Dalai, Fuel, 2012, 96, 77–84 CrossRef CAS.
- T. Toyoda, T. Minami and E. W. Qian, Energy Fuels, 2013, 27, 3769–3777 CrossRef CAS.
- Z. Li, Y. Fu, J. Bao, M. Jiang, T. Hu, T. Liu and Y. N. Xie, Appl. Catal., A, 2001, 220, 21–30 CrossRef CAS.
- D. Li, C. Yang, H. Qi, H. Zhang, W. Li, Y. Sun and B. Zhong, Catal. Commun., 2004, 5, 605–609 CrossRef CAS.
- D. Li, C. Yang, H. Zhang, W. Li, Y. Sun and B. Zhong, Stud. Surf. Sci. Catal., 2004, 147, 391–396 CrossRef CAS.
- H. Qi, D. Li, C. Yang, Y. Ma, W. Li, Y. Sun and B. Zhong, Catal. Commun., 2003, 4, 339–342 CrossRef CAS.
- D. Li, C. Yang, W. Li, Y. Sun and B. Zhong, Top. Catal., 2005, 32, 233–239 CrossRef CAS.
- D. Li, N. Zhao, H. Qi, W. Li, Y. Sun and B. Zhong, Catal. Commun., 2005, 6, 674–678 CrossRef CAS.
- S. Uchiyama, Y. Ohbayashi, T. Hayasaka and N. Kawata, Appl. Catal., 1988, 42, 143–152 CrossRef CAS.
- V. R. Surisetty, A. K. Dalai and J. Kozinski, Appl. Catal., A, 2010, 385, 153–162 CrossRef CAS.
- Y. Okamoto, S. Ishihara, M. Kawano, M. Satoh and T. Kubota, J. Catal., 2003, 217, 12–22 CAS.
- J. Bao, Y.-L. Fu and G.-Z. Bian, Catal. Lett., 2008, 121, 151–157 CrossRef CAS.
- L. Jalowiecki, J. Grimblot and J. P. Bonnelle, J. Catal., 1990, 126, 101–108 CrossRef CAS.
- V. Ponec, Catal. Today, 1992, 12, 227–254 CrossRef CAS.
- H. C. Woo, I. Nam, J. S. Lee, J. S. Chung, K. H. Lee and Y. G. Kim, J. Catal., 1992, 138, 525–535 CrossRef CAS.
- J. Iranmahboob, D. O. Hill and H. Toghiani, Appl. Catal., A, 2002, 231, 99–108 CrossRef CAS.
- B. Yoosuk, D. Tumnantong and P. Prasassarakich, Fuel, 2012, 91, 246–252 CrossRef CAS.
- V. R. Surisetty, Y. Hu, A. K. Dalai and J. Kozinski, Appl. Catal., A, 2011, 392, 166–172 CrossRef CAS.
- G. Leyral, M. Ribes, L. Courthéoux, D. Uzio and A. Pradel, Eur. J. Inorg. Chem., 2012, 4967–4971 CrossRef CAS.
- B. Liu, Y. Chai, Y. Li, A. Wang, Y. Liu and C. Liu, Appl. Catal., A, 2014, 471, 70–79 CrossRef CAS.
- A. Olivas, T. A. Zepeda, I. Villalpando and S. Fuentes, Catal. Commun., 2008, 9, 1317–1328 CrossRef CAS.
- D. Yang, S. J. Sandoval, W. M. R. Divigalpitiya, J. C. Irwin and R. F. Frindt, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 12053–12056 CrossRef CAS.
- D. Yang and R. F. Frindt, J. Mater. Res., 1996, 11, 1733–1738 CrossRef CAS.
- K. S. Liang, R. R. Chianelli, F. Z. Chien and S. C. Moss, J. Non-Cryst. Solids, 1986, 79, 251–273 CrossRef CAS.
- Z.-D. Huang, W. Bensch, L. Kienle, S. Fuentes, G. Alonso and C. Ornelas, Catal. Lett., 2008, 122, 57–67 CrossRef CAS.
- Z.-D. Huang, W. Bensch, L. Kienle, S. Fuentes, G. Alonso and C. Ornelas, Catal. Lett., 2008, 124, 24–33 CrossRef CAS.
- H. C. Woo, J. C. Kim, I. S. Nam, J. S. Lee, J. S. Chung and Y. G. Kim, Appl. Catal., A, 1993, 104, 199–214 CrossRef.
- E. T. Liakakou, E. Heracleous, K. S. Triantafyllidis and A. A. Lemonidou, Appl. Catal., B, 2015, 165, 296–305 CrossRef CAS.
- V. P. Santos, B. Linden, A. Chojecki, G. Budroni, S. Corthals, H. Shibata, G. R. Meima, F. Kapteijn, M. Makkee and J. Gascon, ACS Catal., 2013, 3, 1634–1637 CrossRef CAS.
- M. R. Morrill, N. T. Thao, H. Shou, R. J. Davis, D. G. Barton, D. Ferrari, P. K. Agrawal and C. W. Jones, ACS Catal., 2013, 3, 1665–1675 CrossRef CAS.
- P. Raybaud, J. Hafner, G. Kresse, S. Kasztelan and H. Toulhoa, J. Catal., 2000, 189, 129–146 CrossRef CAS.
- F. Dumeignil, J. F. Paul, E. Veilly, E. W. Qian, A. Ishihara, E. Payen and T. Kabe, Appl. Catal., A, 2005, 289, 51–58 CrossRef CAS.
- Y. Fan, G. Shi, H. Liu and X. Bao, Appl. Catal., B, 2009, 91, 73–82 CrossRef CAS.
- M. Sun, J. Adjaye and A. E. Nelson, Appl. Catal., A, 2004, 263, 131–143 CrossRef CAS.
- F. Labruyere, M. Lacroix, D. Schweich and M. Breysse, J. Catal., 1997, 167, 464–469 CrossRef CAS.
- H. Xiao, D. Li, W. Li and Y. Sun, Fuel Process. Technol., 2010, 91, 383–387 CrossRef CAS.
- P. J. Mangnus, A. Riezebos, A. D. van Langeveld and J. A. Moulijn, J. Catal., 1995, 151, 178–191 CrossRef CAS.
- J. Liu, E. Wang, J. Lv, Z. Li, B. Wang, X. Ma, S. Qin and Q. Sun, Fuel Process. Technol., 2013, 110, 249–257 CrossRef CAS.
- A. Andersen, S. M. Kathmann, M. A. Lilga, K. O. Albrecht, R. T. Hallen and D. Mei, Catal. Commun., 2014, 52, 92–97 CrossRef CAS.
- O. Y. Gutiérez, C. Kaufmann and J. A. Lercher, ChemCatChem, 2011, 3, 1480–1490 CrossRef.
- H. C. Woo, I. S. Nam, J. S. Lee, J. S. Chung and Y. G. Kim, J. Catal., 1993, 142, 672–690 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24406h |
| This journal is © The Royal Society of Chemistry 2016 |