DOI:
10.1039/C6RA24393B
(Paper)
RSC Adv., 2016,
6, 110329-110336
Preparation and model of high-performance shape-memory polyurethane with hydroxylated perylene bisimide†
Received
30th September 2016
, Accepted 5th November 2016
First published on 7th November 2016
Abstract
3,4,9,10-Perylene tetracarboxylic anhydride (PTCDA) was reacted with tris(hydroxymethyl)methyl aminomethane (THAM) to form the corresponding hydroxylated perylene bisimide (PBI). Different weight ratios of PBI were used as the reactive filler with hexamethylene diisocyanate (HDI) and polycaprolactone (PCL) to prepare shape-memory PBI/polyurethane composites (PSMPs). Thermogravimetric analyses were carried out to identify the thermal properties of the PSMPs. The shape-memory behaviors of the PSMPs were characterized by dynamic mechanical analysis (DMA). It was revealed that the PSMP with 5.5 wt% PBI exhibited good shape-memory properties with high shape-fixity ratios (Rf) above 94% and shape-recovery ratios (Rr) above 97%. Additionally, compared to the samples without PBI, the PSMP with 5.5 wt% PBI showed better tensile properties with an elongation at break (εr) of 932.8% and tensile strength (σm) of 52.76 MPa, which were 333.3% and 18.09 MPa higher than those of the samples without PBI, respectively. Furthermore, a three-element model was proposed to discuss the shape-memory behaviors of the PSMPs. The current work is expected to inform ongoing efforts to develop more efficient methods to study the shape-memory behaviors of polymers and has broad implications for the development of shape-memory polyurethanes for engineering applications.
1. Introduction
Shape-memory polymers (SMPs) are a class of smart materials that have the ability to change their shape and recover to their original shape via the application of an external stimulus, such as temperature, pH, humidity, light, magnetism, and electricity.1–10 SMPs along with their inorganic counterparts have drawn considerable attention, giving rise to many interesting developments in both industry and academia over the past decades.11 Above all, the most popular investigated area is thermally induced SMPs.12,13 The shape recovery processes of thermally induced SMPs are well-known: first, a sample is deformed to a temporary shape under an external force at a temperature above the transition temperature (Ttrans, which can be either the glass transition temperature Tg or melting temperature Tm). When the temperature is reduced below Ttrans, the temporary shape can be fixed before the removal of the external force. Once the sample is reheated to above Ttrans, the sample will recover its original shape.2,14,15 SMPs have a variety of applications, including sensors, actuators, high-performance textiles, biomedical devices, packaging materials and self-deployable sun sails in spacecraft.1,2,8,10
Unfortunately, the poor mechanical properties of SMPs limit their potential applications in some areas.16 The preparation of SMPs with fully recoverable and good mechanical properties may promote the development of smart devices and could be combine with other advanced polymer materials.17 Many kinds of polymers have been investigated as SMP materials, such as polyacrylate,18,19 polyurethane20,21 and polynorbornene.22,23 Shape-memory polyurethanes (SMPUs) composed of soft and hard segments are some of the most prominent thermoplastic SMPs.24 Thus, we aimed to synthesize a fully recoverable SMPU with good mechanical properties.
Additionally, micro-phase separation (the formation of hard segment domains and soft segment domains) and the stability of the hard segment domains are the decisive factors that influence the recovery properties.6 Many previous studies on the enhancement of recovery in thermoplastic SMPUs focused on maximizing crystallization in the hard segment11,25 or introducing nano-fillers to reinforce the hard domains via physical attraction with ionic groups,26,27 long alkyl chains,28 or hard segments.29 The thermodynamic incompatibility can be improved to enforce micro-phase separation.
Polycaprolactone glycol (PCL), a semicrystalline polymer, is widely used as the base material for SMPs.6,30,31 However, the poor mechanical properties of PCL limit its potential applications.16 Thus, we wanted to design a reactive filler with special structure and functional groups that can be introduced into SMPUs with PCL to improve the stability of the hard phase and the mechanical properties of the SMPUs.
In a previous work by our research group, the modification of an epoxy composite with 3,4,9,10-perylene tetracarboxylic anhydride (PTCDA) was shown to significantly improve the thermal performance and mechanical properties of the composite.32 Hence, in this study, PTCDA was used as the starting material for a special reactive filler due to its particular chemical structure, which contains a ring structure and anhydride C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. PTCDA reacts with tris(hydroxymethyl)methyl aminomethane (THAM) to form hydroxylated perylene bisimide (PBI). PBI contains six hydroxyl groups, a stiff ring structure, and an imide group; thus, it can be used as a special reactive filler for the preparation of SMPUs. Meanwhile, PBI can be introduced into the hard segments of the SMPUs to improve the hard-phase stability and phase coarsening at the same time. When PBI is used as the hard segment, the strong attraction between PBI and polyurethane is expected to enhance the thermodynamic and mechanical stabilities of shape-memory PBI/polyurethane composites (PSMPs).33 Furthermore, the strong group interaction and stiff ring structure also lead to high thermodynamic incompatibility between the aliphatic soft segments and hard segments, leading to more complete phase separation and further phase coarsening.34 Additionally, a series of corresponding PSMPs were synthesized. The effect of PBI content on the properties of the PSMPs were investigated. Finally, based on the viscoelastic theory of polymers, a three-element model was established to further analyze the shape-memory behaviors of the PSMPs. The model can be used to explain the shape fixation and recovery of shape-memory polymers.
O. PTCDA reacts with tris(hydroxymethyl)methyl aminomethane (THAM) to form hydroxylated perylene bisimide (PBI). PBI contains six hydroxyl groups, a stiff ring structure, and an imide group; thus, it can be used as a special reactive filler for the preparation of SMPUs. Meanwhile, PBI can be introduced into the hard segments of the SMPUs to improve the hard-phase stability and phase coarsening at the same time. When PBI is used as the hard segment, the strong attraction between PBI and polyurethane is expected to enhance the thermodynamic and mechanical stabilities of shape-memory PBI/polyurethane composites (PSMPs).33 Furthermore, the strong group interaction and stiff ring structure also lead to high thermodynamic incompatibility between the aliphatic soft segments and hard segments, leading to more complete phase separation and further phase coarsening.34 Additionally, a series of corresponding PSMPs were synthesized. The effect of PBI content on the properties of the PSMPs were investigated. Finally, based on the viscoelastic theory of polymers, a three-element model was established to further analyze the shape-memory behaviors of the PSMPs. The model can be used to explain the shape fixation and recovery of shape-memory polymers.
2. Experimental
2.1 Materials
PTCDA was obtained from Zhengzhou Alpha Chemical Co. Ltd. (Zhengzhou, China). The solvent N-methyl-2-pyrrolidone (NMP) was analytical grade and provided by Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). THAM was purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Hexamethylene diisocyanate (HDI) and PCL (Mw = 1000) were purchased from Aladdin Reagent Co. Ltd. (Shanghai, China). 4,4′-Biphenol (BP) was obtained from Tokyo Chemical Industry (Shanghai) Development Co., Ltd. The solvent N,N-dimethylformamide (DMF) was analytical grade and provided by Guangdong Xilong Chemical Co. Ltd. (China). The catalyst anhydrous zinc acetate and dibutyltin dilaurate (DBTDL) were chemical grade and provided by Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). DMF and PCL were dried with CaH2 for 24 h and freshly distilled before using.
2.2 Synthesis of PBI
First, 3.92 g PTCDA, 9.68 g THAM, 1.83 g anhydrous zinc acetate and 100 ml NMP were placed into a 250 ml round-bottomed three-neck flask equipped with a nitrogen inlet and a thermometer. The mixture was heated to 80 °C under vigorous stirring and then purged with nitrogen for 15 min. The reaction mixture was then heated to 150–160 °C. After stirring at 150–160 °C for 16 h, the red reaction mixture gradually turned dark-purple. The dark-purple solution was suction-filtered and washed with alcohol to remove the unreacted PTCDA, THAM and anhydrous zinc acetate. The product was dried at 50 °C in a vacuum-drying oven to obtain PBI as a purple powder.
The synthetic route of PBI is shown in Scheme 1.
 |
| | Scheme 1 The synthetic route of PBI. | |
2.3 Preparation of PSMPs
PSMPs were prepared using a two-step polymerization process.35–37 All the PSMPs had the same molar ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5) of PCL to HDI. The mass percent of the PBI to the whole raw materials ranged from 1 to 5.5 wt%. Polyurethane is denoted as PSMP-X, where X indicates the mass percent of the PBI. For example, PSMP-1 means that the preparation of polyurethane uses 1 wt% PBI to the whole raw materials. The reaction was carried out in a round-bottomed three-neck flask equipped with a nitrogen inlet and a thermometer.
:1.5) of PCL to HDI. The mass percent of the PBI to the whole raw materials ranged from 1 to 5.5 wt%. Polyurethane is denoted as PSMP-X, where X indicates the mass percent of the PBI. For example, PSMP-1 means that the preparation of polyurethane uses 1 wt% PBI to the whole raw materials. The reaction was carried out in a round-bottomed three-neck flask equipped with a nitrogen inlet and a thermometer.
As shown in Scheme 2, 2.0 g PCL was first placed into a round-bottom flask and heated to 80 °C under magnetic stirring until it was melted. DMF (10 ml) and DBTDL (0.05 ml) were added to the round-bottom flask. The mixture was then heated to 105 °C for 15 min to remove the moisture and cooled to 80 °C. HDI (0.5 ml) was then added to the moisture-removed mixture. The reaction was kept at 80 °C for 5 h. During the reaction period, 0.025 g PBI was added to 10 ml DMF and with ultrasonic dispersion for 1 h. After the reaction, the BP and the ultrasonically dispersed PBI solution were added to the reacted mixture, which was then magnetically stirred for 12 h at 80 °C under a nitrogen atmosphere to prepare pure shape-memory polyurethane (denoted as PSMP-0) and PSMP-1. Finally, the product was cast into a Teflon plate for further reacting and then dried at 80 °C in an oven for 12 h to obtain film specimens.
 |
| | Scheme 2 Preparation process of PSMP. | |
2.4 Characterization
Fourier transform infrared (FT-IR) spectra were recorded between 4000 and 500 cm−1 on a PerkinElmer 1710 spectrophotometer using KBr pellets at room temperature. A thermogravimetric analysis (TGA) instrument (Netzsch STA-449) was used to measure the thermal stability at temperatures up to 800 °C at a scanning rate of 10 °C min−1. The mechanical properties of the PSMPs were evaluated by tensile measurements. The tensile tests were carried out on a universal tensile tester (type UTM4503BLXY) according to National Standard of China (GB79341-2000) at a strain rate of 5 mm min−1. Field-emission scanning electron microscopy (FE-SEM, JEOL JEM-6610) was used to observe the morphologies of the PSMPs. All samples were cut into specimens with dimensions of 16 mm × 2 mm × 0.3 mm for testing, and at least five specimens were tested for each sample.
Shape-memory effects were investigated by dynamic mechanical analysis (Netzsch, DMA 242C). Shape-memory properties was carried out by cyclic thermomechanical tensile and the main process was: (1) heat a sample to Th and stretch to εm; (2) cool to Tl; (3) remove the load to obtain the temporary strain εu(N); (4) reheat to Th and keep at Th for 5 min to recover the permanent shape εp(N) (N is the number of cycles). The main cycle process is shown in Fig. 3. The shape-memory properties were evaluated based on the shape-recovery ratio (Rr) and shape-fixity ratio (Rf), which were respectively calculated as6
| |
 | (1) |
and
| |
 | (2) |
Five specimens were tested to calculate mean values and standard deviations.
3. Results and discussion
3.1 FTIR analysis
The FT-IR spectra of PTCDA and PBI are shown in Fig. 1. Two characteristic absorption bands at 1752 and 1723 cm−1 were obviously observed in the FT-IR spectrum of PTCDA, which were assigned to the anhydride CO stretching vibration. The band at 3402 cm−1 could be attributed to liquid water. This indicated that PTCDA is liable to absorb moisture. Hence, PTCDA should be heated to 80 °C with nitrogen for 15 min for purging. Upon the reaction of PTCDA with THAM to form PBI, the peaks of the imide CO stretching vibration clearly migrated to 1691 and 1644 cm−1, as seen in the FT-IR spectrum of PBI. The band at 3411 cm−1 could be attributed to the hydroxyl functional groups of PBI, which could react with the isocyanate groups of PCL-based pre-polyurethane. These characteristic absorption bands indicated that PBI was successfully synthesized.
 |
| | Fig. 1 FT-IR spectra of PTCDA and PBI. | |
3.2 Effect of the PBI content on the mechanical properties of PSMPs
As shown in Table 1, the elongation at break (εr) first increased and then decreased and finally increased again with increasing PBI content from 0 to 5.5 wt%. First, the εr value increased from 599.5% to 861.5% when the PBI content increased from 0 to 1.0 wt%. This may be attributed to the hydroxyl groups of PBI, which reacted with the isocyanate groups of PCL-based pre-polyurethane, and these chemically combined bonds could greatly improve the tensile properties of PSMP. When the PBI content further increased to 3.0 wt%, more stress concentrations were formed, causing the εr value of PSMP to drop to 611.1%. As the PBI content continued to increase to 5.5 wt%, the εr value of PSMP rebounded to 932.8%. The large amount of unreacted hydroxyl groups from PBI could form strong hydrogen bonds between each other, which may have partly offset the decline in mechanical properties caused by stress concentration defects. As shown in Table 1, the tensile strength (σm) of PSMP showed the same trend as the aforementioned εr values of PSMP when the PBI content increased. The experimental data revealed that the PSMP with 5.5 wt% PBI showed the best tensile properties, with εr and σm values of 932.8% and 52.76 MPa, respectively. These values were 333.3% and 18.09 MPa higher than the samples without PBI, respectively.
Table 1 Mechanical properties of PSMPs with different weight contents of PBIa
| Sample |
PBI (wt%) |
σm (MPa) |
εr (%) |
| Values in parentheses are standard deviations. |
| PSMP-0 |
0 |
34.67(±5.55) |
599.5(±79.1) |
| PSMP-1 |
1.0 |
44.96(±2.94) |
861.5(±107.8) |
| PSMP-2 |
2.0 |
39.01(±2.40) |
698.6(±175.2) |
| PSMP-3 |
3.0 |
24.56(±7.32) |
611.1(±128.2) |
| PSMP-4 |
4.0 |
40.06(±1.62) |
752.2(±110.6) |
| PSMP-5.5 |
5.5 |
52.76(±2.09) |
932.8(±121.7) |
3.3 Thermal properties of the PSMPs
Fig. 2 shows the thermal decomposition behavior of the PSMPs. The calculated data are listed in Table 2. As shown in Fig. 2, the TGA curves of all samples displayed a one-step degradation mechanism, indicating that addition of PBI did not change the degradation mechanism of the PSMP. The temperature at which a sharp weight loss occurs is an important parameter to demonstrate the thermal stability of PSMP and is denoted as the thermal decomposition temperature. Table 2 shows that PSMPs with PBI display lower thermal decomposition temperatures than the PSMP without PBI, and the thermal decomposition temperature first decreased and then increased with increasing PBI content. For example, the temperatures at a weight loss of 3 wt% for PSMP-0, PSMP-1, PSMP-2, PSMP-3, PSMP-4 and PSMP-5.5 were 277 °C, 270 °C, 269 °C, 269 °C, 263 °C and 270 °C, respectively. This is mainly because PBI has a stiff ring structure and an imide group, which can hinder the formation of an interpenetrating polymer network. When the PSMP was damaged, it required less energy to damage the structure of PSMP. Therefore, the thermal decomposition temperature was reduced. With further increases in PBI content, the large amount of unreacted hydroxyl groups on PBI could form strong hydrogen bonds between each other, which may partly offset the decrease in thermal decomposition temperature due to the steric hindrance effect of the stiff ring structure and imide group.38 The temperature at a weight loss of 3 wt% for PSMP-5.5 rebounded to 270 °C when the PBI content increased to 5.5 wt%. The temperature at a weight loss of 50 wt% of the PSMPs showed a similar trend with increasing PBI content. As shown in Fig. 2 and Table 2, the maximal decomposition temperature also showed the same trend with increasing PBI content as the temperature at a weight loss of 3 wt%.
 |
| | Fig. 2 (a) TGA curves and (b) DTG curves of PSMPs with different PBI contents. | |
Table 2 Thermal stabilities of PSMPs calculated from the TGA and DTG curves
| Sample |
PBI (wt%) |
Temperature at weight loss of 3% (°C) |
Temperature at weight loss of 50% (°C) |
Maximal decomposition temperature (°C) |
Char yield at 550 °C (%) |
| PSMP-0 |
0 |
277 |
332 |
336 |
4.438 |
| PSMP-1 |
1.0 |
270 |
320 |
323.8 |
2.835 |
| PSMP-2 |
2.0 |
269 |
323 |
326.6 |
3.801 |
| PSMP-3 |
3.0 |
269 |
322 |
327.7 |
2.395 |
| PSMP-4 |
4.0 |
263 |
322 |
327.7 |
6.77 |
| PSMP-5.5 |
5.5 |
270 |
331 |
329.1 |
11.33 |
3.4 Shape-memory properties of PSMPs
Fig. 3 shows the representative twice shape memory cycle processes measured by DMA. It is noted that the addition of PBI enhanced not only the mechanical strength, but also the shape-memory properties of PSMP. The Rf and Rr values of the PSMPs were measured by cyclic thermomechanical tensile tests at different temperatures (Netzsch, DMA 242C). The results are shown in Table 3. The optical images of the shape-memory behaviors of the composites are shown in Fig. 1s.† According to previous studies, the key factor in achieving complete recovery was a fully cross-linked network with a large and evenly distributed cross-linked space.6 Herein, the PCL chains were grafted by HDI to form PCL-based pre-polyurethane, and PBI, which worked as a reactive filler, cross-linked the PCL-based pre-polyurethane to form the network structures that allow the full recovery of the PSMP (Fig. 1s†). All the PSMPs exhibited excellent shape memory properties with Rr2 values greater than 94% after two cycles, and the best Rr2 value reached 99.74% (Table 3). The PSMP with 5.5 wt% PBI had outstanding shape-memory properties with Rf2 values greater than 94% and Rr2 values of nearly 98% after two cycles. Although the Rf2 values of PSMP-2, PSMP-3 and PSMP-4 were only around 89%, their Rr2 values were still more than 97% after two cycles. Table 3 also shows that the Rf values increased with increasing PBI content, which was attributed to the larger modulus of the hard segment supplied by PBI. This is because the hard segment with bigger modulus could effectively inhibit the release of internal stress to better fix the temporary shape.39 In addition, the Rf values were not sensitive to the number of cycles, whereas Rr had a higher value in the second cycle than in the first cycle. The main factor affecting Rf was crystallinity, which was affected little by the number of cycles; thus, the Rf values changed little.13 In contrast, the Rr1 values were lower than the Rr2 values, which exceeded 94% after two cycles and reached 99.74% when the PBI content was 4 wt%. The PSMPs may undergo some irreversible segment-chain reorientations and relaxation effects in the first cycle,40 and the effects of these factors wears off or disappears through the first cycles. Thus, the PSMPs had better shape-recovery properties in the second cycle. The Rr1 and Rr2 values were close to each other when the PBI content was 5.5 wt%, indicating that the PSMPs with greater PBI contents had less irreversible segment-chain orientations and relaxation effects owing to the particular structure of PBI.
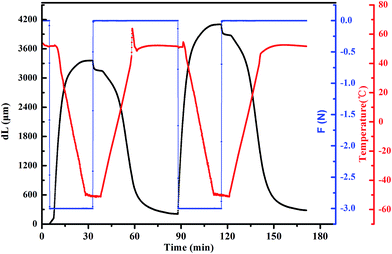 |
| | Fig. 3 Shape-memory cycle process of PSMPs. | |
Table 3 Shape-memory properties of the PSMPs
| Sample |
First cycle |
Second cycle |
| Rf1 (%) |
Rr1 (%) |
Rf2 (%) |
Rr2 (%) |
| PSMP-1 |
86.18 |
63.80 |
85.90 |
94.11 |
| PSMP-2 |
90.69 |
79.22 |
90.40 |
97.63 |
| PSMP-3 |
88.09 |
81.18 |
88.18 |
98.62 |
| PSMP-4 |
89.05 |
87.52 |
88.87 |
99.74 |
| PSMP-5.5 |
93.70 |
93.44 |
94.58 |
97.94 |
3.5 Model building and fitting of PSMPs
A three-element model was established to further study the shape-memory behaviors of the PSMPs. This model is based on the temperature dependence of shape-memory behavior, and the constitutive equation of the three-element mechanical model (Fig. 4) in the one-dimensional form is established by eqn (3):| |
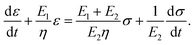 | (3) |
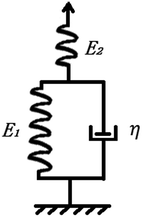 |
| | Fig. 4 Three-element mechanical model. | |
Unlike the ordinary three-element model, the parameters of shape-memory materials (modulus of elasticity, viscosity, relaxation time, etc.) have different values at different temperatures. These temperature dependences are given by the time–temperature equivalence principle:
| |
 | (4) |
| |
 | (5) |
and
| |
 | (6) |
where
E is modulus of elasticity,
ρ is density,
η is viscosity,
a is shift factor of time–temperature equivalence,
C is undetermined constant of time–temperature equivalence,
t is test time,
Ts is the reference temperature, and
τs is the relaxation time at the reference temperature.
The parameters of eqn (3) are related to time because the temperature of the polymer changed over time. According to the time–temperature equivalence principle (Williams–Landel–Ferry equation),
| |
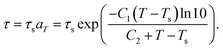 | (7) |
As can be seen from eqn (7), when the temperature of the program is in a constant state, the relaxation time of the SMP will remain the same. However, the relaxation time is different under different temperatures. When the temperature is higher than the switch temperature of the SMP, the chain segments of the SMP start to move, and the relaxation time is short.41 In contrast, when the temperature is lower than the switch temperature of the SMP, the chain segments of the SMP freeze, and the relaxation time is long. Under the condition of stretching and lowering the temperature to freeze the segments, the SMP shape is fixed. As the temperature increases to higher than the switch temperature, the deformed SMP starts to recover. The main steps of the shape-memory process are as follows:42
① At high temperature T1, impose a load σ0.
② Under load σ0, the temperature is lowered from T1 to T0.
③ At temperature T0, remove the load.
④ Raise the temperature to make the polymer shape recover.
Step ④ is the key step in the shape-memory response of an SMP. Step ④ reflects the changes of some important parameters such as shape recovery ratio and shape recovery rate:
| |
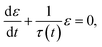 | (8) |
where
t is the load time, and
τ(
t) is determined by
eqn (7), solved
eqn (9):
| |
 | (9) |
Because the polymer heating rate was constant (Ṫ0 = dT/dt), eqn (9) can be changed to eqn (10):
| |
 | (10) |
Therefore, for the three-element model, the fixing ratio (Rf) and recovery ratio (Rr) can be expressed by the following formulae:
| |
 | (11) |
and
| |
 | (12) |
where
t1 is the unloading time, and
t2 is the heating time.
To confirm this three-element model, the numerical simulation results were compared with the experimental results. As shown in Fig. 5a, the numerical and experimental results for PSMP with 5.5 wt% PBI were very similar. Additionally, as shown in in Fig. 5b the simulated and experimental Rf values of PSMPs with different PBI contents were similar, confirming that this model can accurately simulate the shape-memory processes of PSMPs.
 |
| | Fig. 5 (a) Model simulation and experimental results for shape-memory cycles of PSMPs. (b) Model simulation and experimental Rf values for PSMPs. | |
4. Conclusions
In this work, PBI was synthesized, and novel PSMPs were prepared by a two-step polymerization process using PBI as a reactive filler. The effects of PBI on the tensile, thermal and shape-memory properties of the PSMPs were investigated. The results showed that the PSMPs made with PBI had excellent tensile and shape-memory properties. The εr and σm values of the PSMP with 5.5 wt% PBI were 932.8% and 52.76 MPa, respectively, representing respective increases of 55.60% and 52.18% compared to the PSMP without PBI. More importantly, the PSMP with 5.5 wt% PBI exhibited good shape-memory properties with maximum shape-fixity ratio above 94% and shape-recovery ratio above 97%. Additionally, a three-element model was successfully proposed to discuss the shape-memory behaviors of PSMPs.
Acknowledgements
The authors are grateful for the financial support by the National Natural Science Foundation of China (51463007, 51605109 and 51163004), the Innovation Project of Guangxi Graduate Education (YCBZ2015038) and the Guangxi Natural Science Foundation (2015GXNSFBA139231), Guangxi Universities Scientific and Technological Service Project for New Development of City (KY2016YF019).
References
- J. L. Hu, Y. Zhu, H. H. Huang and J. Lu, Prog. Polym. Sci., 2012, 37, 1720–1763 CrossRef CAS
 .
. - M. Behl, M. Y. Razzaq and A. Lendlein, Adv. Mater., 2010, 22, 3388–3410 CrossRef CAS PubMed .
- K. K. Julich-Gruner, C. Lowenberg, A. T. Neffe, M. Behl and A. Lendlein, Macromol. Chem. Phys., 2013, 214, 527–536 CrossRef CAS .
- C. Liu, H. Qin and P. T. Mather, J. Mater. Chem., 2007, 17, 1543–1558 RSC .
- M. Behl and A. Lendlein, Mater. Today, 2007, 10, 20–28 CrossRef CAS .
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 CrossRef CAS .
- H. Meng and G. Q. Li, J. Mater. Chem. A, 2013, 1, 7838–7865 CAS .
- T. Xie, Polymer, 2011, 52, 4985–5000 CrossRef CAS .
- G. J. Berg, M. K. McBride, C. Wang and C. N. Bowman, Polymer, 2014, 55, 5849–5872 CrossRef CAS .
- P. T. Mather, X. F. Luo and I. A. Rousseau, Annu. Rev. Mater. Res., 2009, 39, 445–471 CrossRef CAS .
- D. Ratna and J. Karger-Kocsis, J. Mater. Sci., 2008, 43, 254–269 CrossRef CAS .
- J. S. Leng, X. L. Wu and Y. J. Liu, J. Appl. Polym. Sci., 2009, 114, 2455–2460 CrossRef CAS .
- Y. K. Bai, C. Jiang, Q. H. Wang and T. M. Wang, Carbohydr. Polym., 2013, 96, 522–527 CrossRef CAS PubMed .
- M. Behl and A. Lendlein, Soft Matter, 2007, 3, 58–67 RSC .
- J. S. Leng, X. Lan, Y. J. Liu and S. Y. Du, Prog. Mater. Sci., 2011, 56, 1077–1135 CrossRef CAS .
- L. Xue, S. Y. Dai and Z. Li, Macromolecules, 2009, 42, 964–972 CrossRef CAS .
- W. Voit, T. Ware, R. R. Dasari, P. Smith, L. Danz, D. Simon, S. Barlow, S. R. Marder and K. Gall, Adv. Funct. Mater., 2009, 20, 162–171 CrossRef .
- G. X. Fei, G. Li, L. S. Wu and H. S. Xia, Soft Matter, 2012, 8, 5123–5126 RSC .
- C. M. Yakacki, N. S. Satarkar, K. Gall, R. Likos and J. Z. Hilt, J. Appl. Polym. Sci., 2009, 112, 3166–3176 CrossRef CAS .
- K. Hearon, K. Gall, T. Ware, D. J. Maitland, J. P. Bearinger and T. S. Wilson, J. Appl. Polym. Sci., 2011, 121, 144–153 CrossRef CAS PubMed .
- S. H. Lee, J. W. Kim and B. K. Kim, Smart Mater. Struct., 2004, 13, 1345–1350 CrossRef CAS .
- S. K. Ahn, P. Deshmukh, M. Gopinadhan, C. O. Osuji and R. M. Kasi, ACS Nano, 2011, 5, 3085–3095 CrossRef CAS PubMed .
- D. Yang, D. Y. Gao, C. Zeng, J. S. Jiang and M. R. Xie, React. Funct. Polym., 2011, 71, 1096–1101 CrossRef CAS .
- B. S. Lee, B. C. Chun, Y. C. Chung, K. I. Sul and J. W. Cho, Macromolecules, 2001, 34, 6431–6437 CrossRef CAS .
- W. G. Reyntjens, F. E. Du Prez and E. J. Goethals, Macromol. Rapid Commun., 1999, 20, 251–255 CrossRef CAS .
- S. I. Han, B. H. Gu, K. H. Nam, S. J. Im, S. C. Kim and S. S. Im, Polymer, 2007, 48, 1830–1834 CrossRef CAS .
- Y. Zhu, J. L. Hu, K. W. Yeung, Y. Q. Liu and H. M. Liem, J. Appl. Polym. Sci., 2006, 100, 4603–4613 CrossRef CAS .
- Y. Zhu, J. L. Hu, K. W. Yeung, K. F. Choi, Y. Q. Liu and H. M. Liem, J. Appl. Polym. Sci., 2007, 103, 545–556 CrossRef CAS .
- A. Lendlein, J. Mater. Chem., 2010, 20, 3332–3334 RSC .
- M. Behl, U. Ridder, Y. Feng, S. Kelch and A. Lendlein, Soft Matter, 2009, 5, 676–684 RSC .
- H. Deka, N. Karak, R. D. Kalita and A. K. Buragohain, Carbon, 2010, 48, 2013–2022 CrossRef CAS .
- L. L. Pan, J. F. Ban, S. R. Lu, G. X. Chen, J. Yang, Q. Y. Luo, L. Y. Wu and J. H. Yu, RSC Adv., 2015, 5, 60596–60607 RSC .
- L. S. Zhang, Y. H. Jiang, Z. Xiong, X. Q. Liu, H. N. Na, R. Y. Zhang and J. Zhu, J. Mater. Chem. A, 2013, 1, 3263–3267 CAS .
- Y. J. Li, W. X. Kang, J. O. Stoffer and B. Chu, Macromolecules, 1994, 27, 612–614 CrossRef CAS .
- L. S. T. J. Korley, B. D. Pate, E. L. Thomas and P. T. Hammond, Polymer, 2006, 47, 3073–3082 CrossRef CAS .
- C. Sivakumar and A. S. Nasar, J. Appl. Polym. Sci., 2011, 120, 725–734 CrossRef CAS .
- C. Sivakumar and A. S. Nasar, Eur. Polym. J., 2009, 45, 2329–2337 CrossRef CAS .
- R. Liu and X. D. Wang, Polym. Degrad. Stab., 2009, 94, 617–624 CrossRef CAS .
- L. Sun and W. M. Huang, Soft Matter, 2010, 6, 4403–4406 RSC .
- Y. M. Zhang, Q. H. Wang, C. Wang and T. M. Wang, J. Mater. Chem., 2011, 21, 9073–9078 RSC .
- H. Tobushi, K. Okumura and S. Hayashi, Mech. Mater., 2001, 33, 545–554 CrossRef .
- K. Yu, T. Xie, J. Leng, Y. Ding and H. J. Qi, Soft Matter, 2012, 20, 5687–5695 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24393b |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 


















