DOI:
10.1039/C6RA24283A
(Paper)
RSC Adv., 2016,
6, 104082-104093
Photocatalytic degradation of paraquat dichloride over CeO2-modified TiO2 nanotubes and the optimization of parameters by response surface methodology†
Received
29th September 2016
, Accepted 21st October 2016
First published on 26th October 2016
Abstract
Decontamination of water sources by one-dimensional (1D) nanostructured TiO2 holds great potential due to their unique electronic and textural properties. In this study, CeO2-modified TiO2 nanotubes (Ce–TNTs) have been prepared by impregnation of CeO2 on hydrothermally synthesized TiO2 nanotubes (TNTs). The catalysts were characterized by XRD, HRTEM, EDX, STEM, EELS, DR-UV/VIS spectroscopy and nitrogen adsorption (NA) analyses. The photocatalytic activities of the synthesized Ce–TNTs were examined on the degradation of paraquat dichloride (PQ) under UV light. The modification of TNTs with CeO2 led to an enhancement of the photocatalytic activity. Box–Behnken design (BBD) based on response surface methodology (RSM) was used to optimize three experimental parameters namely; CeO2 ratio, calcination temperature and catalyst loading. ANOVA of the generated quadratic model yielded a coefficient of determination, R2 of 0.9926 and probability, P < 0.0001, which confirms that the model is suitable for predicting the optimum degradation efficiency of PQ. Based on this model, the calcination temperature and CeO2 ratio were the most significant parameters and the interactions between these parameters and the catalyst loading were also significant. The predicted optimum conditions that would give a maximum of 80.798% degradation of PQ in 4 h were 9.01% CeO2 ratio, 760.49 °C calcination temperature and 0.38 g catalyst loading. Validation experiments were conducted in triplicate and an average of 80.27% degradation of PQ was achieved which is in agreement with 80.798% predicted. Under these optimum conditions, TOC analysis showed that 51.10% mineralization of PQ was achieved within 4 h. Therefore, this work further confirms that the photocatalytic treatment of organics-contaminated water can be designed and optimized by RSM.
1. Introduction
Paraquat dichloride (PQ) is a widely used pesticide in agriculture for the treatment of broadleaf and grassy weeds in various types of crops. It is highly toxic to humans and animals and has the potential to cause membrane damage through lipid peroxidation by free radical attack.1 Though, it has been banned in European countries, it still enjoys patronage in many developing countries such that its widespread use and high solubility in water pose a great concern regarding contamination of water sources.2 Thus, it is very essential to treat wastewaters generated as a result of production and application of PQ prior to disposal in order to secure the health of humans, wildlife and the aquatic environment.3 To this effect, advanced oxidation processes (AOPs) which have the capability to completely destroy the PQ to innocuous substances are very much desirable.
Heterogeneous photocatalysis, which is one of the AOPs, is accepted as an efficient, cost-effective and environmentally friendly method of decontaminating water that has been polluted with pesticides and other organic compounds.4–6 TiO2 has gained popularity as the most applied semiconductor in the photocatalytic degradation of organic pollutants due to its availability and cheap cost, chemical stability, photostability, biological inertness and high redox potential.6 Principally, the ability of TiO2 to absorb photons which cause molecular excitations and generate electrons and holes pairs (e−/h+) for redox reactions is responsible for its photocatalytic properties.7 However, TiO2 nanoparticles suffers from low quantum yield as a result of the fast recombination of the photogenerated charge carriers i.e. e−/h+ pairs before they can take part in the redox reactions at the TiO2 surface.8 Thus, 1D nanostructured TiO2 such as TiO2 nanotubes (TNTs) and TiO2 nanorods (TNRs) have been investigated due to the delocalization of electrons and an excellent interconnectivity for charge transfer in these materials which are favourable for preventing rapid e−/h+ recombination.9,10 1D TiO2 have been found to possess higher surface area to volume ratio and higher porosity than their nanoparticles counterpart.11,12 These unique electronic and textural properties accounted for the higher photocatalytic activities reported for 1D TiO2.13
In view of the high porosity of TNTs arising from its hollow structure, it would be more suitable for efficient adsorption of PQ in solution followed by its subsequent degradation. Moreover, because most of the previous studies reported the degradation of PQ over zero-dimensional TiO2 nanoparticles,14–16 TNTs have been selected for this study. TNTs have been synthesized through various methods such as electrochemical anodization,17 sol–gel18 and hydrothermal methods.19 Electrochemical anodization and templated sol–gel method usually produce well-aligned nanotubes but are more complicated because they require the use of templates or substrates and further surface modification of the TiO2 nanotubes obtained will be difficult. On the other hand, hydrothermal method using concentrated NaOH solution is cheaper and easier.20 It does not require any template or substrate and also produces high quality titanate nanotubes with high surface areas and porosity which are subsequently converted to TNTs through calcination. Although, many reports are available about synthesis of TNTs,17–22 only a few works have shown its application for the photocatalytic degradation of pesticides.17,22
Apart from morphological modification to 1D nanostructures, the quantum yield of TiO2 photocatalyst has been improved by doping it with other metallic elements or oxides. Reports have shown that doping of TiO2 nanoparticles with transition metal oxides such as ZnO, NiO, SnO2, WO3, etc. enhanced the charge separation on TiO2 surface and led to higher photocatalytic activities.23–25 Doping of TiO2 nanoparticles with rare earth ions such as Ce and Sm has also been reported to reduce the crystallite size and increase the surface area of TiO2, inhibit its anatase to rutile phase transformation, and prevented the fast recombination of e−/h+ pairs, thus allowing more electrons and holes to participate in the redox reactions leading to the degradation of pollutants.26–28 Among the rare earths, of particular interest is Ce doping due to its f electrons which create the Ce3+/Ce4+ redox couples that facilitate charge separation at the surface of Ce-doped TiO2 thus increasing the quantum yield.26,29 Till date, only a few reports are available about Ce-doped TNTs and the ones found were applied to other processes such as peroxidase mimic30 and photocurrent response analysis.29 To the best of our knowledge, no reports are available on the impregnation of CeO2 on hydrothermally prepared TNTs. Likewise, no report is found on the photocatalytic degradation of PQ over Ce-doped TNTs. Hence, we have studied the photocatalytic degradation of PQ over Ce–TNTs prepared by impregnating CeO2 on TNTs synthesized by hydrothermal method.
To attain the maximum potential of the prepared Ce–TNTs in the photocatalytic degradation of PQ, it is important to optimize the process. The one-factor-at-a-time (OFAT) approach is cumbersome, wasteful in terms of reagent and energy and prone to errors due to its inability to account for the relationship between the interactions of the variables and the response.31–33 Therefore, a more viable approach based on response surface methodology (RSM)34 was adopted. Of the different RSM designs, such as Box–Behnken design (BBD), central composite design (CCD), full factorial design (FFD) and Doehlert design, BBD is regarded as being more economical than others when the same number of variables are involved.35 It requires lesser number of experiments and still gives an excellent prediction of the output.31 Hence, BBD was selected and this is the first report showing the optimization of the photocatalytic degradation of PQ over Ce–TNTs using BBD coupled with RSM. Although, there is a report on the RSM optimization of the degradation of PQ over immobilized TiO2,36 they considered other parameters such as initial PQ concentration, pH, catalyst loading, etc. which were also studied by other researchers with different pollutants,32,33 and the effects of these parameters have been well established. Therefore, this study focused on optimization in terms of the catalytic material considering key parameters viz., (i) calcination temperature which dictates the crystal structure and hence the photocatalytic activity of the catalysts and (ii) the CeO2 ratio which affects charge separation and (iii) the catalyst loading which is also important.
Thus, in this study, Ce–TNTs were synthesized by wet chemical methods (TNTs by hydrothermal method and Ce–TNTs by impregnation method) and applied to the degradation of PQ. The optimization of three important experimental parameters namely; calcination temperature, catalysts loading and CeO2 ratio that affect the degradation efficiency of PQ over the synthesized Ce–TNTs was conducted by RSM coupled with BBD. Linear regression analysis was applied to predict the responses and the predicted responses were compared with experimental values. ANOVA was used to test the fitness of the model for predicting the optimum response. The optimized conditions of independent variables were identified and experiments were performed under the optimized conditions to validate the model prediction. Kinetic and mineralization studies of PQ degradation was carried out under the optimized condition.
2. Experimental
2.1 Materials
Titanium tetraisopropoxide (TTIP), cerium nitrate (Ce(NO3)3·6H2O) and paraquat dichloride (99.9%) were purchased from Sigma-Aldrich. NaOH, HCl, NH4OH and ethanol were obtained from QReC. HNO3 was purchased from EMSURE. Distilled water was used throughout the experiments. All the reagents were used as purchased without further purification.
2.2 Catalyst preparation
TiO2 nanotubes (TNTs) were prepared by hydrothermal method using TiO2 nanoparticles (TNPs) as precursor according to those reported in the literatures except in the synthesis of TNPs.11,18,20,37 Firstly, TiO2 nanoparticles were prepared by sol–gel method as follows: 25 mL of TTIP was dissolved in 50 mL of ethanol. Then, another 35 mL solution containing 2.5 mL of distilled water and 32.5 mL of ethanol was added and stirred for 30 min before the addition of 0.17 mL HNO3. The resultant milky solution was stirred for 2 h and aged at room temperature for 24 h after which it was dried in an oven for 2 days. The TNPs obtained were calcined at 500 °C for 2 h. Then, 0.4 g of the prepared TNPs were mixed with 40 mL of 10 M NaOH solution and transferred into a Teflon-lined autoclave. Hydrothermal treatment was done at 150 °C for 18 h to obtain sodium titanate nanotubes. The titanate nanotubes were washed several times with distilled water and 0.1 M HCl until pH 7 to substitute the Na+ ions with H+ ion. The resultant nanotubes were calcined at 500 °C for 2 h to obtain anatase TNTs.
To prepare Ce–TNTs, anatase TNTs were modified with cerium oxide (CeO2) by impregnation method. Four weight ratios of CeO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TNTs i.e. 5:95, 10:90, 15:85 and 20:80 were prepared. For each CeO2 ratios, the required amount of Ce(NO3)·6H2O (0.098, 0.206, 0.328 and 0.464 g) was dissolved in 10 mL of distilled water and 1 g of anatase TNTs was added to it to give 5, 10, 15 and 20% CeO2 loadings, respectively. This mixture was stirred and left in contact for 30 min after which it was filtered and dried in an oven at 90 °C. The resultant Ce–TNTs were calcined at different temperatures i.e. 500, 600, 700, 800 and 900 °C for 2 h.
:TNTs i.e. 5:95, 10:90, 15:85 and 20:80 were prepared. For each CeO2 ratios, the required amount of Ce(NO3)·6H2O (0.098, 0.206, 0.328 and 0.464 g) was dissolved in 10 mL of distilled water and 1 g of anatase TNTs was added to it to give 5, 10, 15 and 20% CeO2 loadings, respectively. This mixture was stirred and left in contact for 30 min after which it was filtered and dried in an oven at 90 °C. The resultant Ce–TNTs were calcined at different temperatures i.e. 500, 600, 700, 800 and 900 °C for 2 h.
As a reference for EELS analysis, CeO2 was prepared by precipitation method. 12 g Ce(NO3)3·6H2O was 50 mL distilled H2O and stirred for 5 min. Then, NH4OH was added to reach pH 10–11 to obtain the nanoparticles which were washed with distilled H2O and ethanol until pH 7. The nanoparticles were dried and calcined at 500 and 760 °C for 2 h.
2.3 Catalyst characterization
The textural properties of the catalysts were determined by nitrogen adsorption (NA) analysis using micromeritics 3 Flex version 3.01. The samples were outgassed at 350 °C for 240 min at a ramping rate of 10 °C before NA measurement at −196 °C. The crystal structures were determined by X-ray diffraction (XRD) analysis using Siemens X-ray diffractometer, D5000 with CuKα radiation of 0.154 nm at a 2θ range of 5–90°. The morphologies of the Ce–TNTs were determined by atomic resolution scanning/transmission electron microscopy (STEM) using JOEL atomic resolution analytical electron microscope, JEM-ARM 200F. The STEM image was acquired on annular bright field (ABF) detector with 11–20 mrad scattering angle, 40000000× magnification and 0.1 nm probe diameter. The elemental composition was determined by electron dispersive X-ray (EDX) analysis coupled with the HRTEM. In addition, the chemical state of Ti and Ce in the synthesized catalysts were determined by electron energy loss spectroscopy (EELS) available with the STEM instrument. Finally, the optical properties of the catalysts were determined by diffuse reflectance UV/Vis (DR UV-Vis) spectroscopic analysis using UV/VIS spectrometer, Lambda 35, PerkinElmer.
2.4 Photocatalytic degradation experiments
The synthesized Ce–TNTs were applied as photocatalysts in the degradation of modelled PQ polluted water. The photocatalytic degradation experiments were carried out in a photo-reactor equipped with 12 W UV lamp (intensity = 0.6 mW cm−2 and λmax = 365 nm), a flask for holding the pesticide sample and a magnetic stirrer to enhance mass transfer during the photocatalytic process. The intensity of the UV lamp was measured with long wave ultraviolet measuring meter, Blak-Ray model J-221. For each experiment, 250 mL of 15 mg L−1 aqueous PQ solution was placed in the flask and the required amount of catalyst was added. The suspension was covered with a parafilm and a watch glass and stirred in the dark for 1 h to attain adsorption–desorption equilibrium. Then, a 4 mL sample was withdrawn and its UV absorbance was determined and recorded as the initial concentration of PQ against which other concentrations at any time, t, was compared. After this, the suspension was irradiated for 4 h and samples were withdrawn at regular intervals during this period. A blank experiment with similar concentration of PQ but without the addition of catalyst was performed to determine the photolysis of PQ. The degradation was monitored at λmax = 258 nm by UV-Vis spectrophotometer (UV-1601 PC, UV-Visible Spectrophotometer, Shimadzu) and the mineralization was monitored by total organic carbon analyzer (TOC-VE, Shimadzu).
2.5 Experimental design and optimization by RSM
Box–Behnken design (BBD), with 3-factors 3-levels, based on RSM was employed to optimize the photodegradation of PQ over the synthesized Ce–TNTs. For the experimental design, three influential parameters i.e. calcination temperature (°C), catalyst loading (g) and CeO2 ratio (%) were selected as the independent variables. The percentage degradation of PQ was selected as the response, Y (%). The range and levels of the independent variables were selected based on the preliminary results obtained. The independent variables were coded as dimensionless factors; X1, X2 and X3 and their levels and range are shown in Table S1 of the ESI.† Due to the three factors involved, BBD recommended a total of 17 experiments with different combinations of the independent variables. All these experiments were carried out and the results were analysed using the Design Expert software (version 7.1.6 Stat-Ease Inc., USA).32,35,38 The software performed statistical calculations on the experimental data based on 2nd order polynomial model given in eqn (1).| | |
Y (%) = β0 + β1X1 + β2X2 +β3X3 + β11X12 + β22X22 + β33X32 + β12X1X2 +β13X1X3 + β23X2X3
| (1) |
where; Y (%) is the predicted degradation efficiency of PQ, β0 is the interception coefficient, β1, β2, β3 are linear coefficients, β11, β22, β33 are square coefficients, β12, β13, β23 are interaction coefficients, and X1, X2, X3 are independent variables.
Regression analysis was done to determine the relationship between the dependent variable, Y (%) and the independent variables X1, X2 and X3 and the interactions between the independent variables to predict the value of Y (%). Subsequently, the model was tested for its validity using analysis of variance (ANOVA) after which it was used to predict the optimal conditions for the degradation of PQ. Additional experiment based on the RSM-suggested optimum conditions was carried out to verify the accuracy of the model prediction.
3. Results and discussion
3.1 Characterization of catalysts
The crystal structure, surface area and morphology of the catalysts were analysed because they are known to greatly influence the photocatalytic activity of TiO2.20,39 Prior to characterization of catalysts, the photocatalytic screening and RSM optimization studies were performed. The catalysts to be characterized were selected based on the understanding of the variations in their photocatalytic degradation efficiencies under normal and RSM optimized conditions. Therefore, the TNPs used as precursor for TNTs, TNTs calcined at 500 °C used to prepare all the Ce–TNTs and the catalyst with the optimum composition i.e. 9.01% CeO2 ratio and 760 °C calcination temperature were characterized.
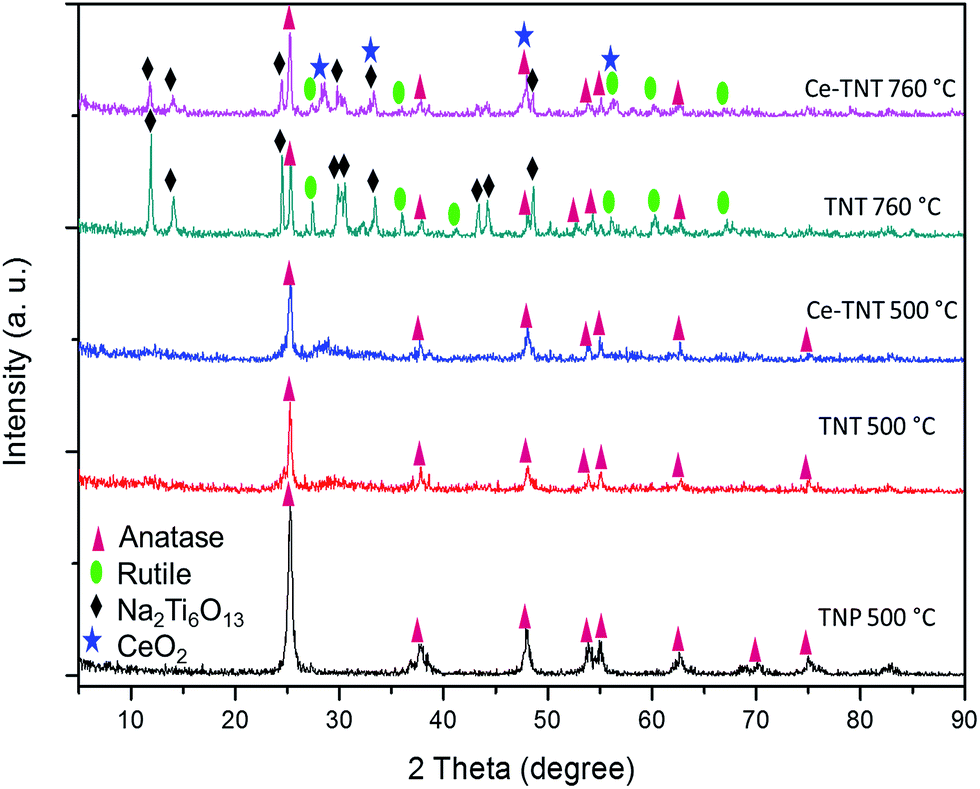
3.1.1 Crystal structure. The XRD spectra of TNPs calcined at 500 °C, TNTs calcined at 500 and 760 °C, and 9% Ce–TNTs calcined at 500 and 760 °C are presented in Fig. 1. It was observed that TNPs, TNTs and Ce–TNTs all calcined at 500 °C showed pure anatase TiO2 peaks (JCPDS: 00-021-1272 and 01-071-1166). This is expected as the phase transition temperature of TiO2 from anatase to rutile is ∼600 °C.40–42 The TNTs and Ce–TNT calcined at 760 °C showed mixed phases of anatase and rutile TiO2 (JCPDS: 01-073-1232). No CeO2 peaks were detected on the Ce–TNTs calcined at 500 °C but weak peaks due to cubic ceria were detected at 760 °C calcination temperature (JCPDS: 00-034-0394). The crystallite size of the catalysts was calculated using the Scherer formula: D = Kλ/βcosθ, where, D is crystallite size, K is Scherer's constant taken as 0.9, λ is X-ray wavelength = 0.154 nm, β is the full width at half maximum (FWHM) in radians and θ is diffraction angle. The effects of CeO2 were seen in lowering the crystallite sizes of Ce–TNTs compared to TNTs as shown in Table S2 of the ESI,† and also in hindering the anatase to rutile phase transition of TiO2 as shown by the higher intensity of rutile peaks in TNTs calcined at 760 °C compared to Ce–TNTs calcined at the same temperature (Fig. 1). This agrees well with the findings of other researchers.26–28 Due to the fact that the ionic radius of Ce4+, 1.02 Å is larger than that of Ti4+, 0.68 Å, most of the Ce4+ ions will be located at the grain boundaries.27 This can also be linked to the suppression of the crystal growth of the undesirable Na2Ti6O13 species in Ce–TNTs compared with TNTs, both calcined at 760 °C. The Na2Ti6O13 species resulted from the incomplete removal of Na+ ions during the washing process and are not photocatalytically active.11
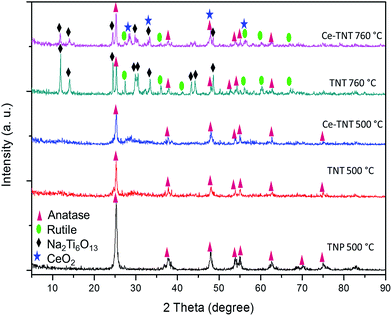 |
| | Fig. 1 XRD spectra of catalysts calcined at 500 and 760 °C for 2 h. | |
3.1.2 Textural properties. The adsorption–desorption isotherms and pore size distribution of the catalysts are presented in Fig. S1.† It can be observed in Fig. S1a† that all the catalysts exhibit the type IV isotherm according to IUPAC classification,43 although TNTs calcined at 500 °C and Ce–TNTs calcined at 500 and 700 °C seem to be a mixture of types IV and V isotherms. Hysteresis loops were found in all the synthesized catalysts which shows that they are mesoporous materials.20,44 However, there were variations in the type of hysteresis loops exhibited by TNPs and TNTs. TNPs showed the type H2 hysteresis loop which implies the existence of ink bottle-shaped pores while TNTs and Ce–TNTs showed type H3 hysteresis loop typical of slit-shaped pores.45 Other researchers have also reported the type H3 hysteresis for the hydrothermally synthesized TNTs.39 From the pore size distribution presented in Fig. S1b,† it can be observed that all the catalysts showed a fairly narrow pore size distribution. Table 1 shows the surface areas (measured by the Brunauer–Emmet–Teller (BET) method), pore size (measured by Barrett–Joyner–Halenda (BJH) method) and pore volumes (also measured by BJH method) and the XRD crystallite sizes of the various catalysts. The synthesized TNTs have higher surface areas than TNPs which is similar to the findings of other researchers.13,46 The surface area decreased as the calcination temperature increased.47 This is due to the increase in crystallite size and widening of pores. However, it is noteworthy that the catalysts retained their mesoporous properties even at high calcination temperatures which is desirable for efficient surface interaction with the pollutants.48
Table 1 Textural properties of catalysts
| Catalyst |
Surface areaa (m2 g−1) |
Pore volumeb (cm3 g−1) |
Pore diameterb (nm) |
XRD crystallite sizec (nm) |
| Measured by BET method. Measured by BJH method. Calculated using Scherer formula, —: means not measured. |
| TNP 500 °C |
11.87 |
0.03 |
5.37 |
23.51 |
| As-prepared TNT |
142.49 |
0.29 |
6.34 |
— |
| TNT 500 °C |
49.53 |
0.31 |
19.24 |
27.43 |
| Ce–TNTs 500 °C |
36.49 |
0.22 |
19.1 |
20.58 |
| Ce–TNTs 760 °C |
10.89 |
0.13 |
46.88 |
32.91 |
3.1.3 Morphology. The HRTEM images of the 9.01% Ce–TNTs calcined at 500 and 760 °C are presented in Fig. 2. It was observed in Fig. 2a that at 500 °C calcination temperature, the Ce–TNTs consisted of multi-wall nanotubes with average inner diameter of 5.00 nm, average outer diameter of 10.18 nm and length 143 nm. As the calcination temperature increased to 760 °C (Fig. 2c), the outer diameter of the nanotubes increased to 36.51 nm with a corresponding increase in length to around 600 nm and the loss of the inner hole. This may account for the reduction of surface area observed in Fig. S1 of the ESI.† The spherical nanoparticles seen on the TNTs surface in Fig. 2a and c were envisaged to be the impregnated CeO2 nanoparticles. This was confirmed by the measured lattice spacing, d of 0.311 nm corresponding to the (111) plane of cubic ceria (Fig. 2b) obtained from the profile of fast Fourier transform (FFT) shown in Fig. S2 of the ESI.† Furthermore, EDX spectra in Fig. S3 and S4 of the ESI† and the EDX mapping images in Fig. 2f and g confirm that the spherical nanoparticles observed on the TNTs are CeO2. The STEM image in Fig. 2d shows the interface between CeO2 and TNTs and its FFT is shown in Fig. S5.† An atomic resolution STEM micrograph (Fig. 2e) taken from the marked area in Fig. 2d shows the lattice orientation of all the elements presents in Ce–TNTs calcined at 500 °C as a representative for all the other prepared Ce–TNTs. The annular bright field (ABF) detector used to acquire the STEM image in Fig. 2e is more sensitive to low atomic number elements, hence lighter element appear brighter. Visibly, oxygen appear brighter than the cerium, titanium and sodium. Cropped-out image denotes the atomic spacing measurement for each element.
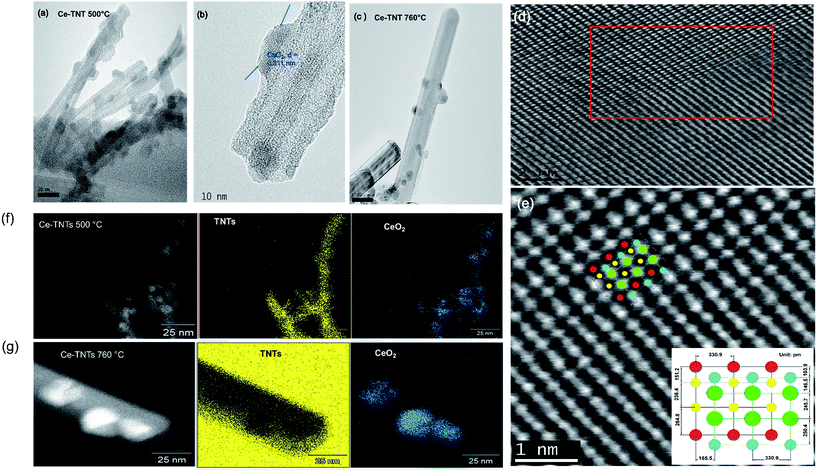 |
| | Fig. 2 HRTEM images of (a) Ce–TNTs calcined at 500 °C, (b) single Ce–TNT calcined at 500 °C on which the lattice spacing of CeO2 has been measured as indicated by the arrows, (c) Ce–TNTs calcined at 760 °C (d) STEM image of Ce–TNTs calcined at 500 °C showing the interface between CeO2 and TNTs, (e) STEM image taken from marked area in (d), (Ce: , Ti: , Ti: , Na: , Na: and O: and O: ), (f) EDX mapping of 9.01% Ce–TNTs calcined at 500 °C and (g) EDX mapping of 9.01% Ce–TNTs calcined at 760 °C. ), (f) EDX mapping of 9.01% Ce–TNTs calcined at 500 °C and (g) EDX mapping of 9.01% Ce–TNTs calcined at 760 °C. | |
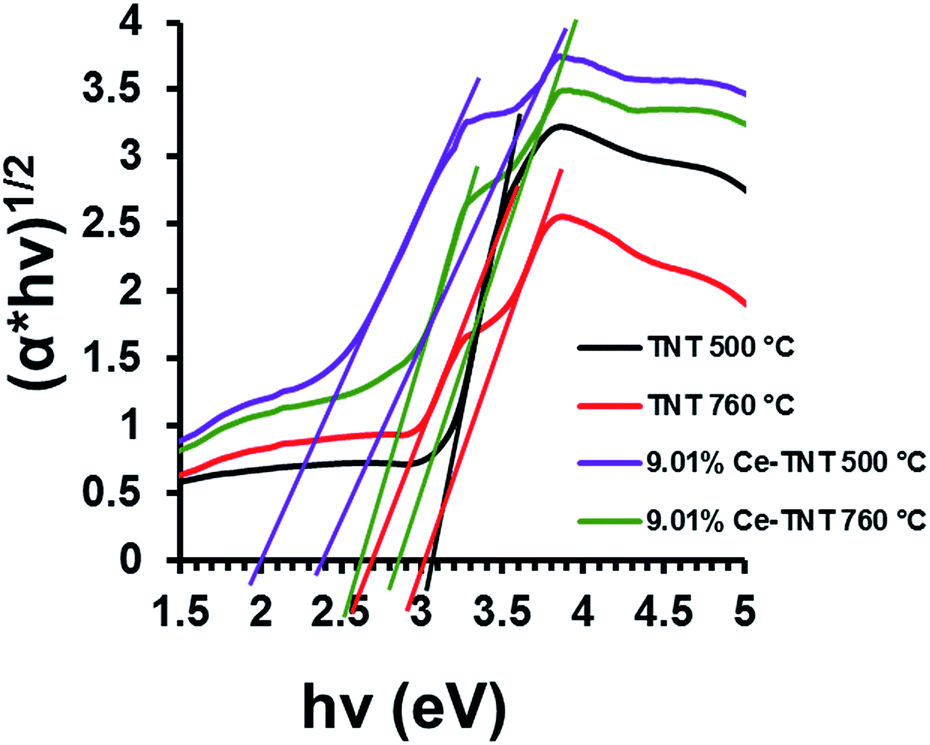
3.1.4 Optical properties. Fig. 3 shows the calculated band gap energies of TNTs and Ce–TNTs determined from the DR UV-Vis spectra. The reflectance spectra were converted to Kubelka–Munk function which was subsequently applied to the data using the equation (αhν)1/2 = A(hν − Ebg) where, α is the absorption coefficient, hν is the photon energy, Ebg is the band gap energy and A is a constant relative to the material. From the Tauc plot in Fig. 3, the band gap energies were determined by extrapolating the linear portion of the graph to the hν axis and are 3.05 eV and 3.00/2.70 eV for TNTs calcined at 500 and 760 °C and 2.00/2.40 eV and 2.60/2.85 eV for Ce–TNTs calcined at 500, and 760 °C, respectively. The TNTs calcined at 760 °C exhibited two band gaps which may be due to the mixed phases of anatase and rutile TNTs in this catalyst, the lower for rule and the higher energy for anatase.49,50 Two band gaps were also observed on Ce–TNTs samples. Band gap lowering observed in Ce–TNTs may be attributed to the 4f transitions of Ce3+/Ce4+ on the surface of TiO2.51 Other researchers have also reported band gap lowering for CeO2-modified TiO2.26,51
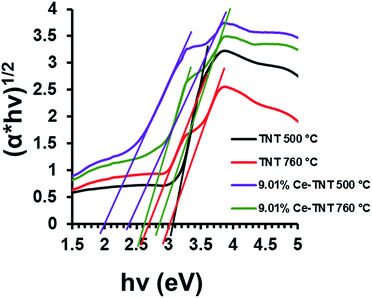 |
| | Fig. 3 Band gap energy of TNTs and 9.01% Ce–TNTs. | |
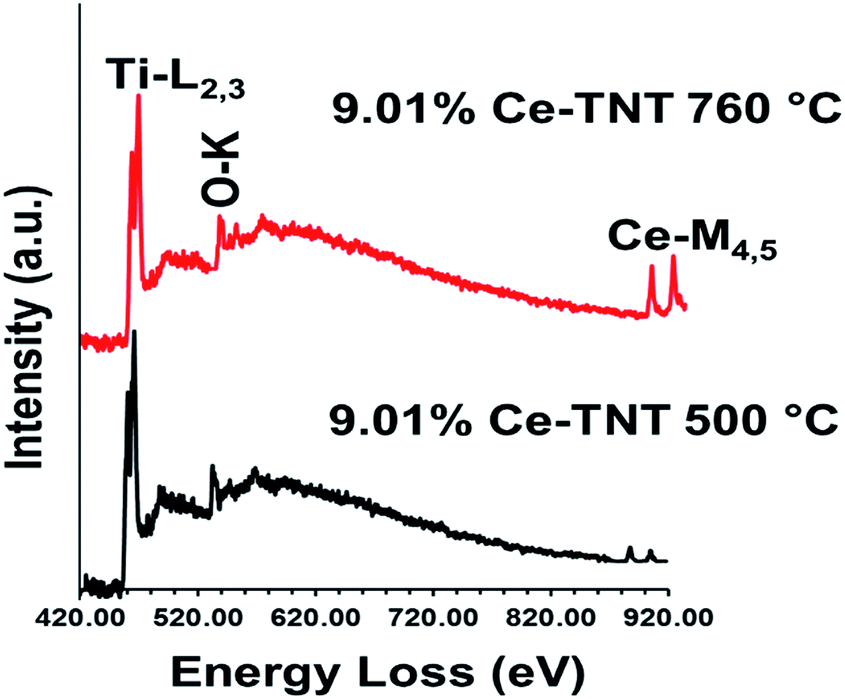
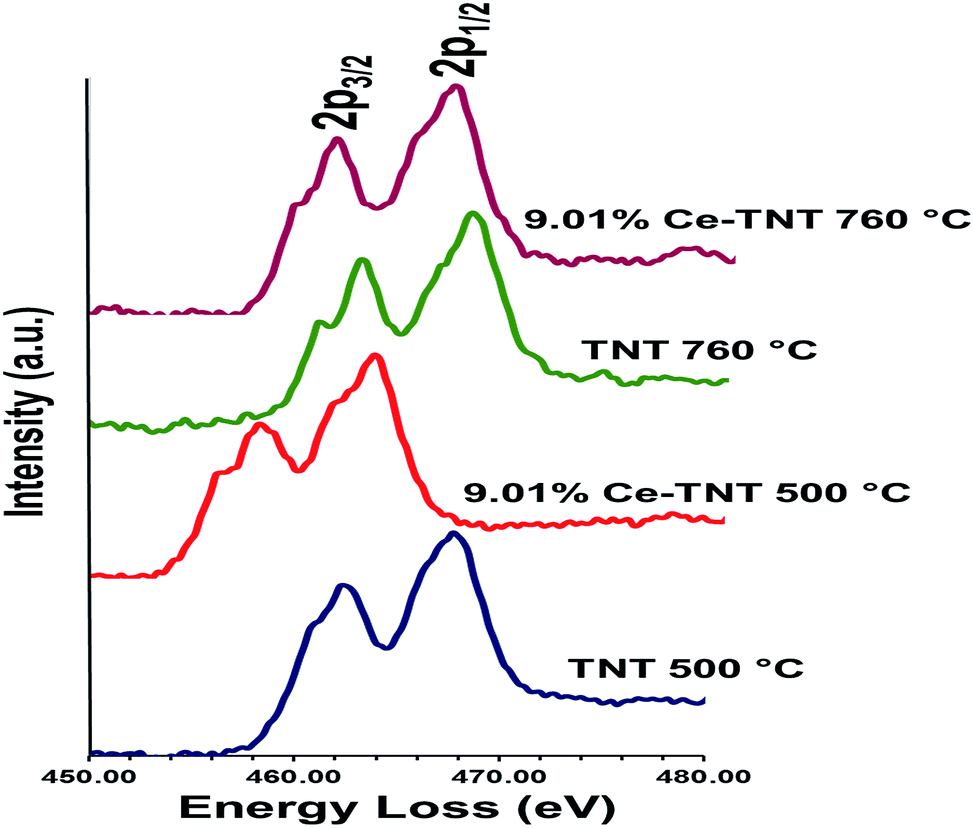
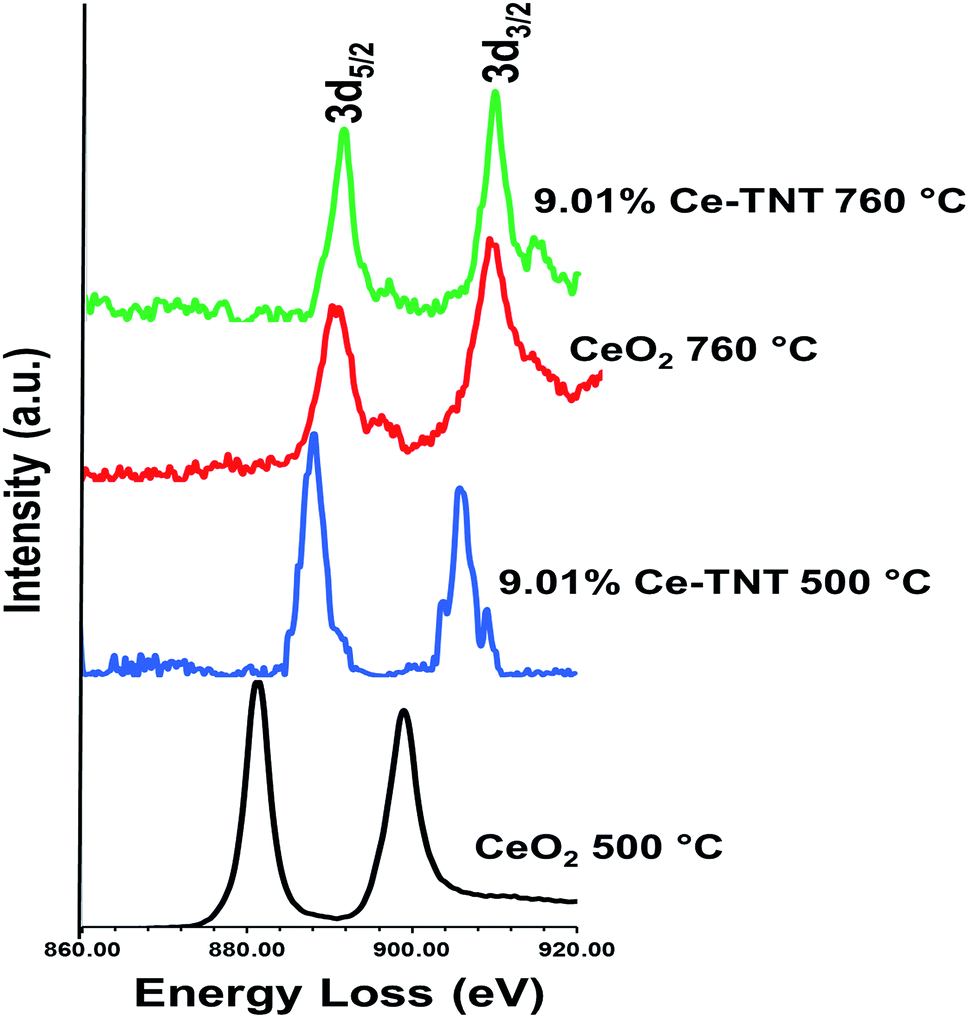
3.1.5 Chemical state. Fig. 4 shows the EELS spectra of 9.01% Ce–TNTs calcined at 500 and 760 °C. The EELS spectra of TNTs and CeO2 calcined at 500 and 760 °C used as references for EELS measurement of Ce–TNTs are also presented in Fig. S6 and S7 of the ESI.† The white lines of O-K, Ti-L2,3 and Ce-M4,5 are clearly visible in these spectra. The white line intensity ratios L3/L2 or M5/M4 have been used to determine the valence states of the Ti and Ce, respectively, in this study, since they vary with d-electron occupancy.52 It is reported that an increase in M5/M4 ratio indicated an increase in Ce3+ content.53 Hence, the reversal of the intensity of M45 lines observed in CeO2 calcined at 500 °C indicates high Ce3+ concentrations.54 The Ti-L2,3 spectra of TNTs references and the synthesized Ce–TNTs are shown in Fig. 5. The ratios of Ti L3/L2 white lines are 0.775, 0.783, 0.690 and 0.772 for TNTs 500 °C, TNTs 760 °C, Ce–TNTs 500 °C and Ce–TNTs 760 °C, respectively. The peaks of modified and unmodified TNTs looks similar, hence the possibility of existence of Ti3+ is ruled out. For the Ce M5/M4, the intensity ratios for CeO2 calcined at 500 and 760 °C and Ce–TNTs calcined at 500 and 760 °C shown in Fig. 6 are 1.129, 0.750, 1.089 and 0.869, respectively. It was observed that the peaks in Ce–TNTs calcined at 500 °C exhibited reversal of the intensity of M4,5 and are at a lower energy than that calcined at 760 °C. The high M5/M4 intensity ratio in Ce–TNTs 500 °C and the higher M5/M4 ratio of Ce–TNTs 760 °C compared to CeO2 760 °C suggest that the Ce–TNTs samples are rich in Ce3+.53,54
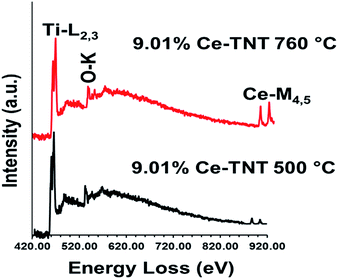 |
| | Fig. 4 EELS spectra of 9.01% Ce–TNTs calcined at 500 and 760 °C. | |
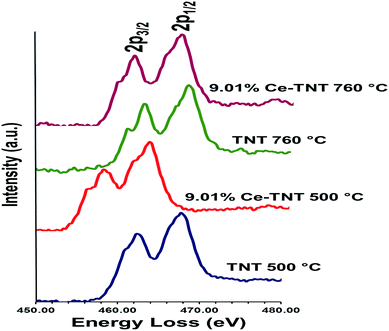 |
| | Fig. 5 EELS spectra of Ti-L2,3 in TNTs and Ce–TNTs. | |
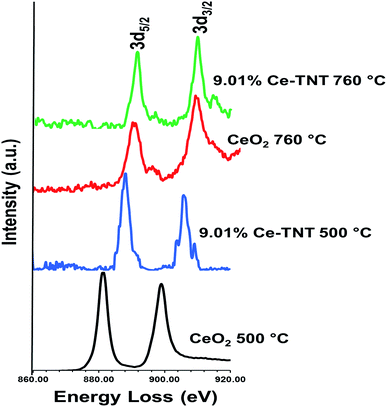 |
| | Fig. 6 EELS spectra of Ce-M4,5 in TNTs and Ce–TNTs. | |
3.2 Preliminary photocatalytic degradation experiments
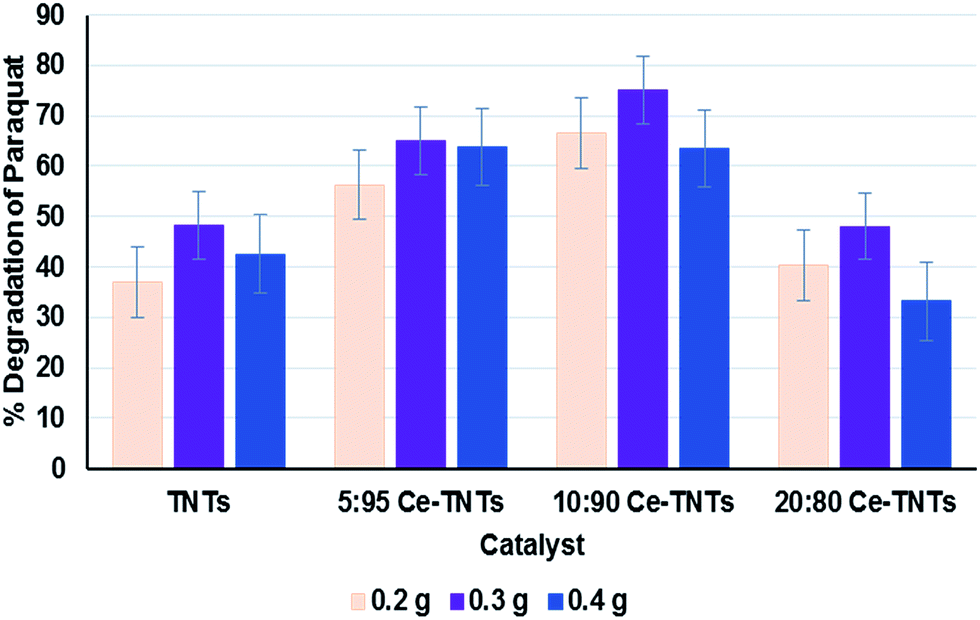
Prior to optimization, preliminary experiments were performed to give an insight into the photocatalytic properties of the synthesized TNTs and Ce–TNTs. The photolysis of PQ was found to be negligible, only 0.03% after 4 h of irradiation which is in agreement with the findings of other researchers.1,16 However, the presence of catalysts in the reaction system resulted in a substantial degradation of PQ. Fig. 7 shows the degradation efficiency of PQ over the synthesized TNTs and Ce–TNTs with different CeO2 ratios and different calcination temperatures using 0.2 g of catalysts. The results indicate that there was a great improvement in the degradation rate of PQ over Ce–TNTs compared to unmodiffied TNTs. This may be explained in terms of the interfacial charge transfer taking place between TNTs and the C3+/C4+ redox couples of CeO2 species which prevented rapid recombination of the e−/h+ pairs, thus leading to an increase in quantum yield.26 The amount of CeO2 loaded on TNTs also affected the performance of the Ce–TNTs in PQ degradation. The degradation efficiency initially increased with an increase in CeO2 ratio from 5% to 10% and decreased with further increase of CeO2 ratio to 20%. This may be because too high CeO2 loading will result in high accumulation of CeO2 at the grain boundaries which will result in a lowering of crystallinity and this amorphous ceria will act as sites for rapid e−/h+ pairs recombination.26,55 In addition, the calcination temperature played a vital role in determining the photocatalytic activities of the catalysts. This is because it dictates the phases of TiO2 present and the crystallinity of the catalyst as shown in the XRD spectra in Fig. 1. At low calcination temperature of 500 °C, the catalysts consisted of only anatase TiO2 compared to those calcined above 700 °C consisting of mixed phases of anatase and rutile TiO2 (Fig. 1). It had been reported that when anatase and rutile TiO2 co-exist, they create a synergistic effect by prolonging the life span of charge carriers due to the differences in their electrode potentials.56 Moreover, the crystallinity of the catalysts increased with an increase in calcination temperature and high crystallinity is desirable for preventing e−/h+ pairs recombination.57,58 Furthermore, XRD peaks due to CeO2 species were only visible at high calcination temperature, which implies higher crystallinity and better ability to trap electrons from TNTs to prevent rapid e−/h+ pairs recombination. Too high calcination at 900 °C resulted in total loss af anatase phase and hence a lower activity. The photocatalytic activities of all the TNTs and Ce–TNTs were lower than expected from their 1D morphology and mesoporosity. This is because some of the TiO2 crystals exist as Na2Ti6O13 (Fig. 1) which is not photocatalytically active, thereby reducing the active sites that take part in the degradation of PQ. Fig. 8 shows the effect of catalyst loading on the degradation rate of PQ at a constant calcination temperature of 700 °C. The degradation efficiency increased with an increase in catalyst loading from 0.2 g to 0.3 g for all the TNTs and Ce–TNTs. However, a further increase of the catalyst loading to 0.4 g resulted in a decrease in the degradation efficiency. Usually, an increase in catalyst loading up to an optimum amount will increase the rate of redox reactions after which aggregation of particles will result in loss of active sites and hinder efficient light penetration through the irradiated solution.32,58,59 These observations showed that there exists an optimum value for each of the studied variables which were subsequently determined by RSM.
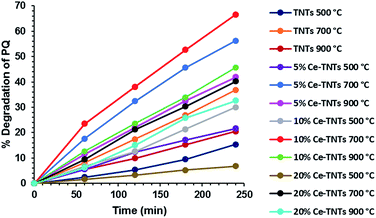 |
| | Fig. 7 Photocatalytic degradation studies of PQ showing the effect of CeO2 ratio and calcination temperature on the degradation efficiency; catalyst loading was 0.2 g. | |
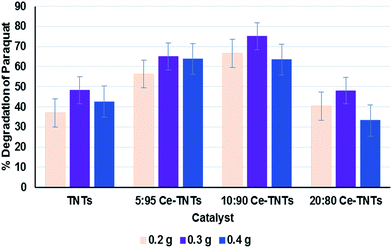 |
| | Fig. 8 Effect of catalyst loading on the degradation efficiency of PQ; calcination temperature was 700 °C. | |
3.3 RSM model fitting and analysis of the response surface
The 17 experiments recommended by BBD of RSM were performed and the experimental and predicted results are presented in Table 2. The empirical second order polynomial equation derived from the experimental data and used to predict the degradation rate of PQ (Y%) is given in eqn (2).| | |
Y = 71.25 + 22.18X1 + 1.62X2 − 5.76X3 + 5.77X1X2 + 2.97X1X3 − 4.48X2X3 − 22.85X12 − 0.93X22 − 0.18X32
| (2) |
where, Y is the degradation efficiency of PQ while X1, X2 and X3 are calcination temperature, catalyst loading and CeO2 ratio, respectively.
Table 2 Experimental design variables with the predicted and actual values of % paraquat dichloride degradationa
| Run |
Independent variable |
% Degradation, Y |
| X1 |
X2 |
X3 |
Experimental |
Predicted |
| X1: calcination temperature, X2: catalyst loading, X3: CeO2 ratio. |
| 1 |
700 |
0.2 |
15 |
59.45 |
59.24 |
| 2 |
600 |
0.3 |
15 |
7.96 |
9.32 |
| 3 |
700 |
0.3 |
10 |
72.11 |
71.25 |
| 4 |
700 |
0.3 |
10 |
68.29 |
71.25 |
| 5 |
700 |
0.2 |
5 |
61.74 |
61.80 |
| 6 |
600 |
0.3 |
5 |
25.67 |
26.76 |
| 7 |
600 |
0.4 |
10 |
22.43 |
21.13 |
| 8 |
800 |
0.3 |
15 |
60.70 |
59.61 |
| 9 |
800 |
0.3 |
5 |
66.55 |
65.19 |
| 10 |
700 |
0.3 |
10 |
76.26 |
71.25 |
| 11 |
700 |
0.4 |
15 |
53.58 |
53.52 |
| 12 |
700 |
0.3 |
10 |
70.59 |
71.25 |
| 13 |
800 |
0.4 |
10 |
75.89 |
77.04 |
| 14 |
700 |
0.4 |
5 |
73.78 |
73.99 |
| 15 |
700 |
0.3 |
10 |
68.98 |
71.25 |
| 16 |
600 |
0.2 |
10 |
30.59 |
29.44 |
| 17 |
800 |
0.2 |
10 |
60.95 |
62.25 |
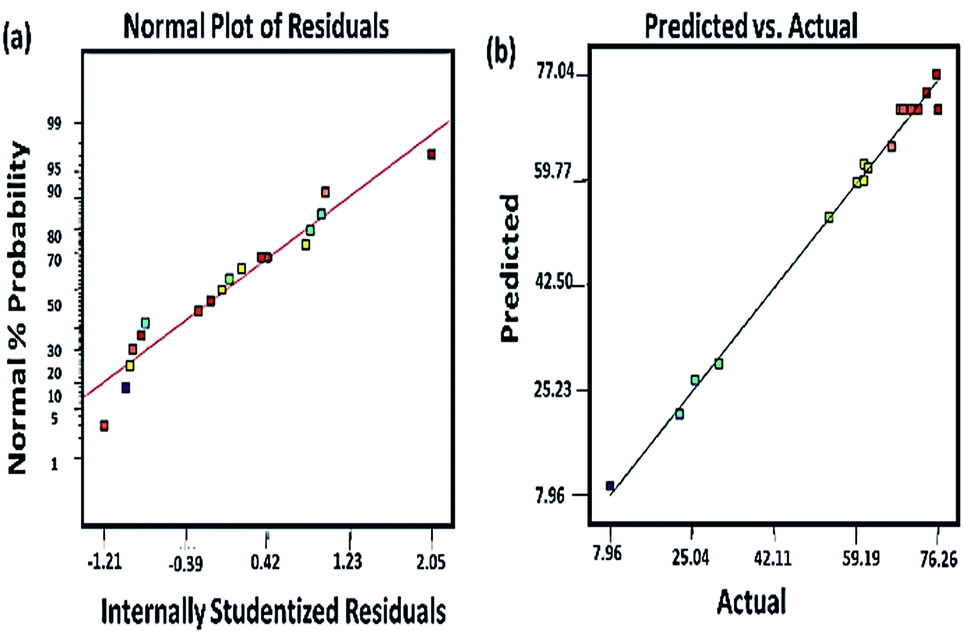
3.3.1 Model fitting. For the model to be adopted, it is necessary to verify its appropriateness. This was carried out by ANOVA. The ANOVA supported by Design Expert software and the data is presented in Table 3. From Table 3, the model F value of 104.97 and probability, P value of <0.0001 indicates that the model is significant and there is 99.99% confidence that it can be used to predict the optimal response of the PQ degradation over the synthesized Ce–TNTs. Probability, P-values less than 0.05 indicate that the model terms are significant and values greater than 0.10 indicate that the model terms are not significant. Therefore, in this case, the calcination temperature (X1) and CeO2 ratio (X3) have much significant influence on the degradation of PQ than the catalyst loading. In terms of the interactions of the independent variables, the interaction of calcination temperature and catalyst loading and that of catalyst loading and CeO2 ratio are all significant. Moreover, lack of fit test is essential to check the adequacy of a model for predicting the optimum response. In this case, the F-value for lack of fit is very low, 0.4, indicating that the lack of fit is not significant relative to the pure error and the model can well be adopted for optimizing the degradation of PQ over Ce–TNTs catalyst. Furthermore, the regression coefficients were utilized to correlate the similarity between the predicted response and the experimental values obtained. From this, R2 of 0.9926 was obtained which is in reasonable agreement with the adjusted R2 of 0.9832 and the predicted R2 of 0.9638. Generally, R2 values greater than 0.80 is desirable60 and thus this model is considered appropriate for optimizing the degradation of PQ over Ce–TNTs within the selected experimental range. In addition, the plots of residuals (Fig. 9a) and predicted versus actual experimental values (Fig. 9b) further confirmed that the experimental results are well fitted to the model prediction with all the data points falling close to the straight lines. The plots of other residuals which also support the validity of the model are given in Fig. S8 of the ESI.†
Table 3 ANOVA for the response surface quadratic model for paraquat dichloride degradationa
| Source |
Regression coefficient |
F-Value |
P-Value |
| R2 = 0.9926, adjusted R2 = 0.9832 and predicted R2 = 0.9638. |
| Model |
|
104.97 |
<0.0001 |
| X1, calcination temperature |
22.18 |
525.91 |
<0.0001 |
| X2, catalyst loading |
1.62 |
2.80 |
0.1381 |
| X3, CeO2 ratio |
−5.76 |
35.42 |
0.0006 |
| X1X2 |
5.77 |
17.83 |
0.0039 |
| X1X3 |
2.97 |
4.70 |
0.0668 |
| X2X3 |
−4.48 |
10.72 |
0.0136 |
| X12 |
−22.85 |
293.75 |
<0.0001 |
| X22 |
−0.93 |
0.49 |
0.5072 |
| X32 |
−8.18 |
37.62 |
0.0005 |
| Lack of fit |
|
0.4 |
0.7584 |
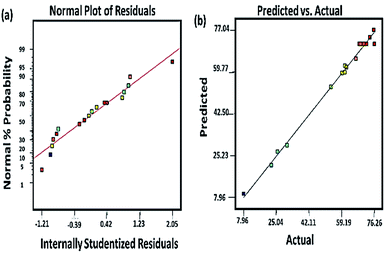 |
| | Fig. 9 (a) The plot of normal probability against internally studentized residuals and (b) the plot of the RSM predicted response value versus experimentally observed values in the photocatalytic degradation of PQ. | |
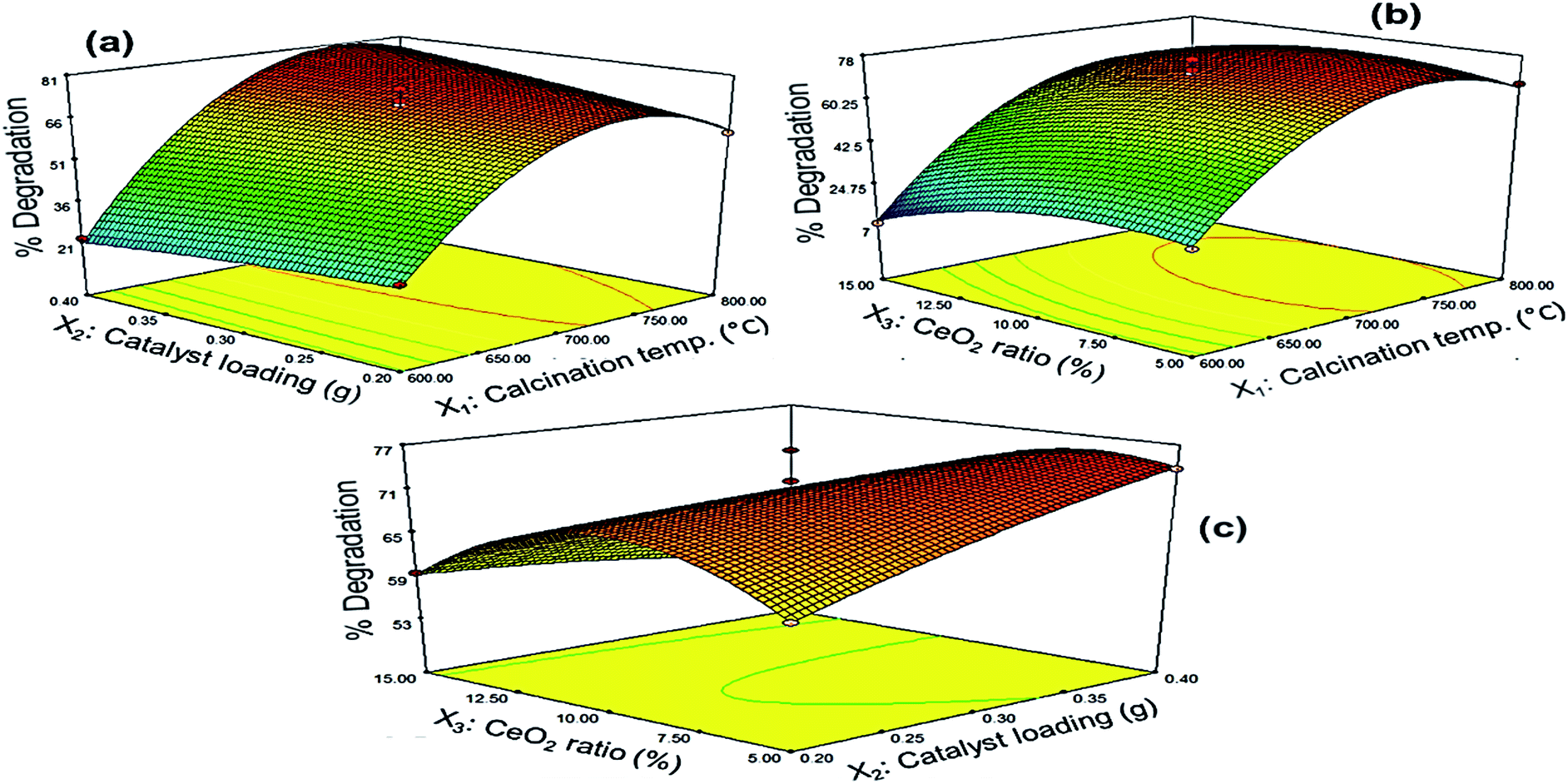
3.3.2 Response surface analysis. The three-dimensional response surfaces of PQ degradation over Ce–TNTs are given in Fig. 10 and the corresponding 2D contour plots are given in Figs. S9–S11 of the ESI.† Fig. 10a shows the effect of the interaction between calcination temperature and catalyst loading on the degradation efficiency of PQ. It was observed that high calcination temperature from 700 °C upwards favoured efficient degradation of PQ. Below this temperature, increasing the catalyst loading did not lead to a remarkable improvement of the degradation efficiency. Above 700 °C, the degradation rate increased with an increase in both calcination temperature and catalyst loading upto an optimum value slightly above 750 °C after which it decreased. This may be explained in terms of the mesoporosity, crystallinity and composition of the catalysts. At low calcination temperature of 600–690 °C, the catalysts have high surface areas but their crystallinity is low compared to when calcined above 700 °C. In heterogeneous catalysis, we know that high surface area leads to higher rate of reaction. But for an efficient photocatalytic process, there must be a compromise between crystallinity and surface area such that the catalyst must have both reasonable surface area and appropriate crystallinity.56 This is because the amorphousness of catalyst will interfere with the efficient utilization of supplied photons by inducing rapid recombination of charge carriers.56,57 In the present case, although the surface area of the catalysts suffered serious reduction at high calcination temperatures, the catalysts are still mesoporous (Fig. S1†) such that they can still effectively adsorb the PQ in solution to bring about its degradation. Furthermore, the high mesoporous structure of Ce–TNTs calcined at 760 °C favours enhanced absorption of applied photons, leading to improved photocatalytic activity.48,61 Moreover, the presence of mixed phases of anatase and rutile TiO2 above 700 °C and the higher crystallinity of CeO2 species contributed to the higher photocatalytic activities at high temperatures between 700 and 800 °C. The above explanations may be the reason why increasing the catalyst loading at low calcination temperature did not bring about any significant improvement in the degradation efficiency of PQ as compared to that observed at high calcination temperatures. A decline in degradation rate as a result of high catalyst loading may be due to the scattering of light by the excess catalyst which led to poor utilization of the supplied photons and hence low quantum yield.32 Likewise, the effect of the interaction between CeO2 ratio and calcination temperature on the degradation efficiency is shown in Fig. 10b. It can be observed that a very high CeO2 ratio above 12% is not desirable at low calcination temperatures as explained earlier for the preliminary experiments. A pronounced interaction effect was only observed above 700 °C when the percentage degradation increased with an increase in both calcination temperature and CeO2 ratio upto 775 °C and 11.5%, respectively, after which a decline in degradation efficiency was observed. This can also be explained interms of the crystallinity because the peaks due to CeO2 species were only visible in the XRD spectra (Fig. 1) at high calcination temperatures. Hence, the CeO2 species is more photocatalytically active at high calcination temperatures. After the CeO2 species exceeded the optimum amount, an excess amount might have created a negative effect by acting as sites for rapid e−/h+ recombination. Nevertheless, Fig. 10c shows the interaction between CeO2 ratio and catalyst loading. It was observed that a combination of very low CeO2 ratio and very low catalyst loading or very high CeO2 ratio and very high catalyst loading resulted in low degradation efficiency. Rather, a combination of high CeO2 ratio and low catalyst loading and vice versa favoured high degradation efficiency. This implies that at a low CeO2 ratio, more catalyst was needed in the reaction system before the effect of the CeO2 species could be felt but at high CeO2 ratio, less amount of catalyst was sufficient to exert the effect of the CeO2 species on the degradation efficiency of PQ. All the observations from the response surface are in agreement with the preliminary results earlier discussed.
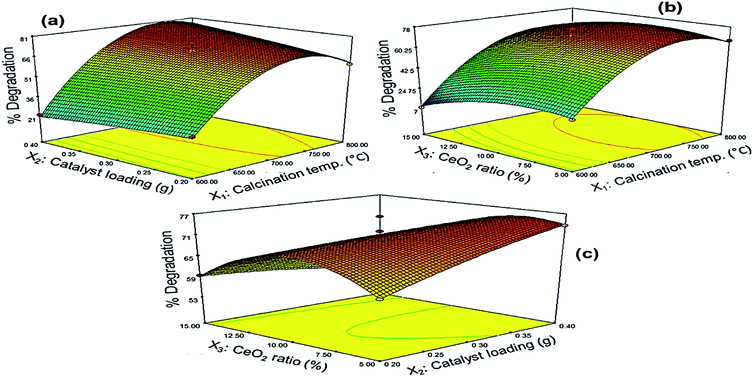 |
| | Fig. 10 Response surfaces of the effects of interactions between (a) calcination temperature and catalyst loading, (b) calcination temperature and CeO2 ratio and (c) catalyst loading and CeO2 ratio, on the degradation efficiency of PQ. | |
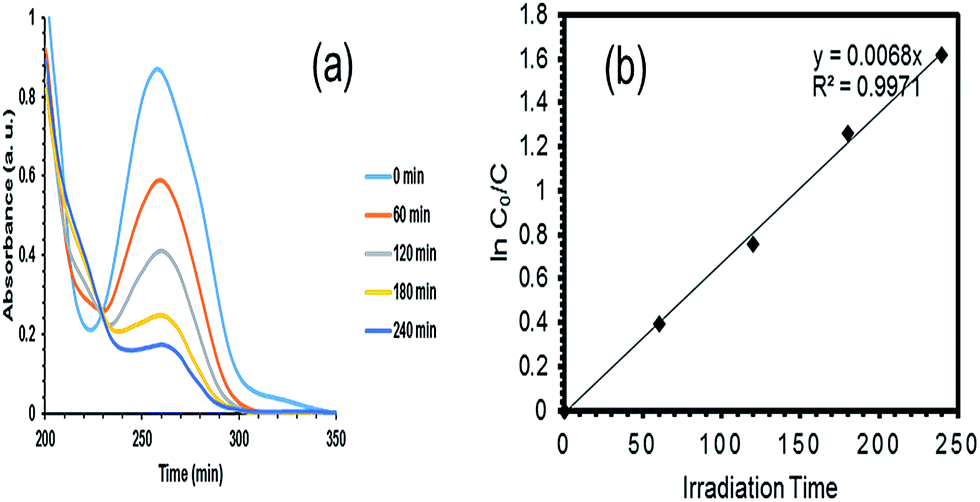
3.3.3 Optimization and validation. After the model was confirmed fit, the optimization function of the Design Expert software was used to determine the optimum values of the independent variables to achieve the highest degradation of PQ over the synthesized Ce–TNTs. The independent variables were kept in range while the response (% degradation) was set to be maximized. The software predicted that 80.798% degradation would be obtained if CeO2 ratio of 9.01%, calcination temperature of 760.49 °C, and catalyst loading of 0.38 g were employed to degrade 250 mL of 15 g L−1 PQ aqueous solution. This prediction was verified by conducting additional experiments in triplicate and the average value obtained was 80.27% which is in agreement with the RSM predicted value. This confirms the validity of the model for predicting the highest degradation efficiency of PQ over the Ce–TNTs photocatalyst. The UV spectra showing the rate of disappearance of PQ with time is given in Fig. 11a while the plot of its degradation efficiency with time is given in Fig. S12 of the ESI.†
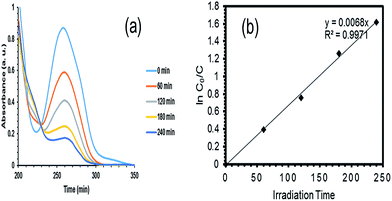 |
| | Fig. 11 (a) UV-Vis spectrum and (b) kinetics of the degradation of PQ during 4 h of UV irradiation under the optimized conditions (9.01% CeO2 ratio, 760.49 °C calcination temperature and 0.38 g catalyst loading). | |
3.4 Kinetics of the degradation of PQ under optimized conditions
The RSM optimized condition was adopted to study the kinetics of the degradation of PQ over Ce–TNTs. In most cases, the degradation of organic pollutants were reported to follow the Langmuir–Hinshelwood (L–H) first order kinetic model.62 However, for dilute systems, with concentrations far less than 10−3 M, the L–H model is simplified to the apparent first order kinetic model:
| lnC0/C = kKt = kappt |
where, C0 is the initial concentration after adsorption and equilibration, C is the concentration at any time, t, k is the rate constant which is the slope of the plot of lnC0/C versus time, K is the equilibrium constant and kapp is the apparent rate constant.63
In this study, the concentration of PQ used was 5.83 × 10−5 M. Therefore, the degradation kinetics shown in Fig. 11b fitted well with the L–H apparent first order kinetics with apparent rate constant, kapp of 6.8 × 10−3 min−1, half life, t1/2 of 102 min and R2 of 0.9971.
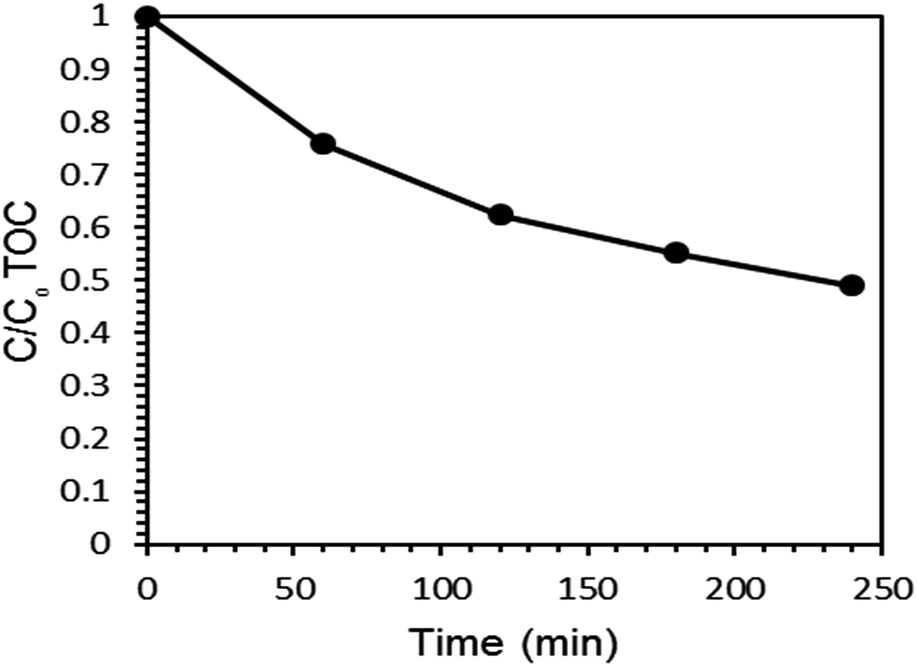
3.5 Mineralization of PQ under optimized conditions
Total organic carbon (TOC) analysis was employed to check the rate of mineralization of PQ over the Ce–TNTs under optimized condition. The rate of mineralization of PQ with time is given in Fig. 12. A total of 51.10% mineralization was obtained within 4 h of irradiation. It was observed that TOC removal was slower than the degradation of PQ. This is in agreement with the observations of other researchers too.64,65 The difference between the degradation and mineralization can be attributed to the existence of intermediate products. The main intermediates in PQ degradation were identified as monopyridone, dipyridone and 4-carboxymethyl-1-methylpyridinium ion14,16 which were further completely mineralized to CO2, H2O, and inorganic ions after 12 h of UV (125 W high pressure mercury lamp) irradiation.16
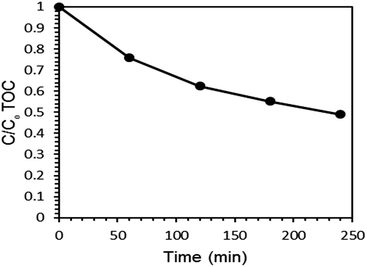 |
| | Fig. 12 TOC removal in the mineralization of PQ during 4 h of UV irradiation. | |
4. Conclusions
An investigation on the photocatalytic activity of CeO2-modified TiO2 nanotubes (Ce–TNTs) on the degradation of paraquat dichloride (PQ) was successfully carried out in this study. Ce doping enhanced charge separation on TNTs surface, reduced the crystallite size and preserved the presence of mixed phases of anatase and rutile TiO2 at high calcination temperature above 700 °C. This accounted for the higher photocatalytic activities of the Ce–TNTs. The Box–Behnken design (BBD) of the response surface methodology (RSM) was utilized to optimize the photocatalytic degradation of PQ over the synthesized Ce–TNTs. The model prediction of RSM fitted well with the experimental data with R2 and Adjusted R2 of 0.9926 and 0.9832, respectively. Overall, RSM predicted the optimum conditions which will give 80.798% degradation of PQ as 0.38 g catalyst loading, 760.49 °C calcination temperature and 9.01% CeO2 ratio. Experiments were performed to validate the RSM suggested conditions. 80.27% degradation and 51.10% mineralization of PQ was obtained under the optimum conditions which proved that RSM can be used to optimize the degradation and mineralization of PQ and other organic pollutants over the synthesized Ce–TNTs. The synthesized Ce–TNTs has a strong potential to mitigate the impacts of pesticides and other organic pollutants on our water sources.
Acknowledgements
The authors express their gratitude to Universiti Teknologi Malaysia (UTM) and the Ministry of Higher Education, Malaysia (MOHE) for funding this research under the FRGS grant (4F770) and N. A. Eleburuike sincerely thank the Organization for Women in Science for the Developing World (OWSD) and Swedish International Development Corporation (SIDA) for her PhD fellowship.
References
- M. I. Cantavenera, I. Catanzaro, V. Loddo, L. Palmisano and G. Sciandrello, J. Photochem. Photobiol., A, 2007, 185, 277–282 CrossRef CAS.
- C. Oliveira, M. S. F. Santos, F. J. M. HÓdar, G. Schaule, A. Alves and L. M. Madeira, Chem. Eng. J., 2012, 210, 339–349 CrossRef CAS.
- S. Chiron, A. Fernandez-Alba, A. Ridriguez and E. Garcia-Calvo, Water Res., 2000, 34, 366–377 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- C. Guillard, J. Disdier, J. M. Herrmann, C. Lehaut, T. Chopin, S. Malato and J. Blanco, Catal. Today, 1999, 54, 217–228 CrossRef CAS.
- A. Fujishima, T. Rao and D. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- S. Singh, H. Mahalingam and P. K. Singh, Appl. Catal., A, 2013, 462–463, 178–195 CrossRef CAS.
- S. Dong, J. Feng, M. Fan, V. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- G. Melcarne, L. Marco, E. Carlino, F. Martina, M. Manca, R. Cingolani, G. Gigli and G. Ciccarella, J. Mater. Chem., 2010, 20, 7248–7254 RSC.
- Q. Lu, Z. Lu, Y. Lu, L. Lv, Y. Ning, H. Yu, Y. Hou and Y. Yin, Nano Lett., 2013, 13, 5698–5702 CrossRef CAS PubMed.
- M. Qamar, C. R. Yoon, H. J. Oh, N. H. Lee, K. Park, D. H. Kim, K. S. Lee, W. J. Lee and S. J. Kim, Catal. Today, 2008, 131, 3–14 CrossRef CAS.
- C. Tsai and H. Teng, Chem. Mater., 2004, 16, 4352–4358 CrossRef CAS.
- W. Liu, J. Gao, F. Zhang and G. Zhang, Mater. Trans., 2007, 48, 2464–2466 CrossRef CAS.
- E. Moctezuma, E. Leyva, E. Monreal, N. Villegas and D. Infante, Chemosphere, 1999, 39, 511–517 CrossRef CAS.
- R. Ali and S. H. Hassan, Malaysian J Anal Sci, 2008, 12, 77–87 Search PubMed.
- M. H. Florêncio, E. Pires, A. L. Castro, M. R. Nunes, C. Borges and F. M. Costa, Chemosphere, 2004, 55, 345–355 CrossRef PubMed.
- C. B. D. Merian, T. Cottineau, D. Robert and P. Drogui, Appl. Catal., B, 2016, 19, 1–6 CrossRef.
- J. Qiu, W. Yu, X. Gao and X. Li, Nanotechnology, 2007, 18, 295604 CrossRef.
- J. Cabrera, H. Alarcón, A. López, R. Candal, D. Acosta and J. Rodriguez, Water Sci. Technol., 2014, 70, 972–979 CrossRef CAS PubMed.
- J. Yu, H. H. Yu, B. Cheng, X. Zhao and Q. Zhang, J. Photochem. Photobiol., A, 2006, 182, 121–127 CrossRef CAS.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef CAS.
- H. Yu, X. Wang, H. Sun and M. Huo, J. Hazard. Mater., 2010, 184, 753–758 CrossRef CAS PubMed.
- R. Nadarajan, W. A. W. A. Bakar, R. Ali and R. Ismail, J. Taiwan Inst. Chem. Eng, 2016, 64, 106–115 CrossRef CAS.
- K. Salehi, B. Shahmoradi, A. Bahmani, M. Pirsaheb and H. P. Shivaraju, Desalin. Water Treat., 2016, 1–11 Search PubMed.
- N. Azri, W. A. W. A. Bakar and R. Ali, J. Taiwan Inst. Chem. Eng., 2016, 62, 283–296 CrossRef CAS.
- L. Matějová, K. Kočí, M. Reli, L. Čapek, A. Hospodková, P. Peikertová, Z. Matěj, L. Obalová, A. Wach, P. Kuśtrowski and A. Kotarba, Appl. Catal., B, 2014, 152–153, 172–183 CrossRef.
- R. Verma, S. K. Samdarshi and J. Singh, J. Phys. Chem. C, 2015, 119, 23899–23909 CAS.
- C. H. Liang, F. B. Li, C. S. Liu, J. L. Lü and X. G. Wang, Dyes Pigm., 2008, 76, 477–484 CrossRef CAS.
- Y. Tan, S. Zhang and K. Liange, Nanoscale Res. Lett., 2014, 9, 67–72 CrossRef PubMed.
- H. Zhao, Y. Dong, P. Jiang, G. Wan and J. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 6451–6461 CAS.
- M. Bezerra, R. Santelli, E. Oliveira, L. Villar and L. Escaleira, Talanta, 2008, 76, 965–977 CrossRef CAS PubMed.
- T. Sharma, A. P. Toor and A. Rakor, RSC Adv., 2015, 5, 25059–25065 RSC.
- M. Vaez, A. Z. Moghaddam and S. Alijani, Ind. Eng. Chem. Res., 2012, 51, 4199–4207 CrossRef CAS.
- R. H. Myers, D. C. Montgomery and C. M. Anderson-Cook, Response surface methodology: process and Product optimization using designed experiments, John Wiley & Sons Inc., Hoboken, New Jersey, 4th edn, 2015 Search PubMed.
- G. Wang, S. Zhang, T. Li, X. Xu, Q. Zhong, Y. Chen, O. Deng and Y. Li, RSC Adv., 2015, 5, 58010–58018 RSC.
- P. Tantriratna, W. Wirojanagud, S. Neramittagapong, K. Wantala and N. Grisdanurak, Indian J. Chem. Technol., 2011, 18, 363–371 CAS.
- L. Wei, H. Wang, Z. Wang, M. Yu and S. Chen, RSC Adv., 2015, 5, 74347–74352 RSC.
- P. Sharma, L. Singh and N. J. Dilbaghi, J. Hazard. Mater., 2009, 164, 1024–1029 CrossRef CAS PubMed.
- S. Song, J. Tu, Z. He, F. Hong, W. Liu and J. Chen, Appl. Catal., A, 2010, 378, 169–174 CrossRef CAS.
- N. T. Nolan, M. K. Seery and S. C. Pillai, J. Phys. Chem. C, 2009, 113, 16151–16157 CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- D. J. Reidy, J. D. Holmes and M. A. Morris, J. Eur. Ceram. Soc., 2006, 26, 1527–1534 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1982, 54, 2201–2218 CrossRef.
- Y. H. Tan, J. A. Davis, K. Fujikawa, N. V. Ganesh, A. V. Demchenko and K. J. Stine, J. Mater. Chem., 2012, 22, 6733–6745 RSC.
- G. Leofanti, M. Padovani, G. Tozzola and B. Venturelli, Catal. Today, 1998, 41, 207–219 CrossRef CAS.
- W. Zhou, H. Liu, R. I. Boughton, G. Du, J. Lin, J. Wang and D. Liu, J. Mater. Chem., 2010, 20, 5993–6008 RSC.
- P. GÓrska, A. Zaleska, E. Kowalska, T. Klimczuk, J. W. Sobczak, E. Skwarek, W. Janusz and J. Hupka, Appl. Catal., B, 2008, 84, 440–447 CrossRef.
- B. Fang, Y. Xing, A. Bonakdarpour, S. Zhang and D. P. Wilkinson, ACS Sustainable Chem. Eng., 2015, 3, 2381–2388 CrossRef CAS.
- D. O. Scanlon, C. M. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- V. Pfeifer, P. Erhart, S. Li, K. Rachut, J. Morasch, J. Brötz, P. Reckers, T. Mayer, S. Rühle, A. Zaban, I. M. Seró, J. Bisquert, W. Jaegermann and A. Klein, J. Phys. Chem. Lett., 2013, 4, 4182–4187 CrossRef CAS.
- J. F. de Lima, M. H. Harunsani, D. J. Martin, D. Kong, P. W. Dunne, D. Gianolio, R. J. Kashtiban, J. Sloan, O. A. Serra, J. Tang and R. I. Walton, J. Mater. Chem. A, 2015, 3, 9890–9898 Search PubMed.
- H. K. Schmid and W. Mader, Micron, 2006, 37, 426–432 CrossRef CAS PubMed.
- A. M. D'Angelo, A. C. Y. Liu and A. L. Chaffe, J. Phys. Chem. C, 2016, 120, 14382–14389 Search PubMed.
- P. Gao, Z. Wang, W. Fu, Z. Liao, K. Liu, W. Wang, X. Bai and E. Wang, Micron, 2010, 41, 301–305 CrossRef CAS PubMed.
- C. Wei, X. Tang, J. Liang and S. Tan, J. Environ. Sci., 2007, 19, 90–96 CrossRef CAS.
- Q. Zhang, L. Gao and J. Guo, Appl. Catal., B, 2000, 26, 207–215 CrossRef CAS.
- J. Yu, H. Yu, B. Cheng, X. Zhao, J. C. Yu and W. Ho, J. Phys. Chem. B, 2003, 107, 13871–13879 CrossRef CAS.
- C. Sahoo and A. K. Gupta, J. Environ. Sci. Health, Part A, 2013, 48, 694–705 CrossRef CAS PubMed.
- W. Jiang, J. A. Jeons, D. D. Dionysiou and K. E. O'Shea, J. Photochem. Photobiol., A, 2013, 262, 7–13 CrossRef CAS.
- J. Zhang, D. Fu, Y. Xu and C. Liu, J. Environ. Sci., 2010, 22, 1281–1289 CrossRef CAS.
- B. Fang, A. Bonakdarpour, K. Reilly, Y. Xing, F. Taghipour and D. P. Wilkinson, ACS Appl. Mater. Interfaces, 2014, 15488–15498 CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS.
- N. A. Ramos-Delgado, M. A. Gracia-Pinilla, L. Maya-Treviño, L. Hinojosa-Reyes, J. L. Guzman-Mar and A. Hernández-Ramírez, J. Hazard. Mater., 2013, 263, 36–44 CrossRef CAS PubMed.
- G. Echavia, F. Matzusawa and N. Negishi, Chemosphere, 2009, 76, 595–600 CrossRef CAS PubMed.
- I. Konstantinou, T. Sakellarides, V. Sakkas and T. Albanis, Environ. Sci. Technol., 2001, 35, 398–405 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra24283a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Rusmidah Alia and
Muhammad Firdaus Omarb
*a,
Rusmidah Alia and
Muhammad Firdaus Omarb

 , Ti:
, Ti: , Na:
, Na: and O:
and O: ), (f) EDX mapping of 9.01% Ce–TNTs calcined at 500 °C and (g) EDX mapping of 9.01% Ce–TNTs calcined at 760 °C.
), (f) EDX mapping of 9.01% Ce–TNTs calcined at 500 °C and (g) EDX mapping of 9.01% Ce–TNTs calcined at 760 °C.