
Design and fabrication of graphene fibers based on intermolecular forces and charge properties in a novel acidic system
Abstract
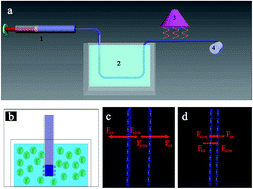
Graphene is one of the most famous carbon-based materials. Integrating two dimension (2D) graphene nanoflakes to macroscopic one dimension (1D) fibers could enrich graphene-based materials and its applications. This paper describes a new strategy of fabricating macroscopic graphene oxide fibers based on intermolecular forces and their charge properties in a new acidic coagulation system. The formation mechanism of graphene oxide fibers was proposed. The graphene fibers were prepared by following chemical reduction. The resultant graphene fibers have strong strain of 107.9 MPa and outstanding electrochemical properties. The cyclic voltammetry and galvanostatic charge–discharge studies for graphene fibers exhibited typical capacitive behavior with good rate stability. This work extended the methods for preparation of graphene fibers. The prepared macroscopic 1D graphene fibers may have a wide range of applications such as supercapacitors, sensors, smart and conductive textiles, etc.
 Please wait while we load your content...
Please wait while we load your content...

