
Mechanisms of reactions of Ru(III)-based drug NAMI-A and its aquated products with DNA purine bases: a DFT study†
Pramod Kumar Shah,
Kanika Bhattacharjee and
Pradeep Kumar Shukla *
*
Department of Physics, Assam University, Silchar–788011, India. E-mail: pkshukla@ymail.com; pradeep.kumar.shukla@aus.ac.in
First published on 29th November 2016
Abstract
It is believed that Ru(III)-based drugs including NAMI-A and KP1019, which have entered clinical trials, are hydrolysed readily in vivo to form a number of more reactive aquated complexes which then react with the biological targets. However, no conclusive report on this matter is available in the literature. Therefore, the mechanisms of reactions of NAMI-A and its mono- and di-aquated products (in which one/two chloride ligands are replaced by one/two water molecules) at the N7 site of guanine as well as the reaction of mono-aquated NAMI-A at the N7 site of adenine have been investigated using density functional theory. The present contribution provides the detailed structural properties of reactant complexes (RCs), transition states (TSs) and product complexes (PCs) involved in these reactions obtained at the M06-2X/(LanL2DZ+6-31G**) level of theory in the gas phase as well as the energetics and rate constants obtained at the M06-2X/(LanL2DZ+6-311+G**) level of theory in the gas phase and aqueous media. The solvent effect of aqueous media was estimated using the conductor-like polarisable continuum model (CPCM). It is found that both the mono- and di-aquated products of NAMI-A are more reactive than NAMI-A itself toward the N7 site of guanine. It is further found that the mono-aquated NAMI-A reacts with the N7 site of adenine more favourably than with the N7 site of guanine, the barrier energy and rate constant of its reaction with adenine (guanine) being 22.67 (27.15) kcal mol−1 and 1.46 × 10−4 s−1 (7.55 × 10−8) in aqueous media. It shows that NAMI-A is first hydrolysed to form more reactive aquated complexes which then react with the biological targets like adenine and guanine. Thus, the present study is expected to be helpful in understanding the mechanisms of analogues of NAMI-A, and other Ru-based drugs also.
1. Introduction
Although the platinum-based drugs like cisplatin are widely used in the treatment of many types of cancer including testicular, ovarian and bladder cancer, these drugs suffer from several drawbacks, such as limited solubility, dose-limiting side effects and intrinsic or acquired resistance in some cancer types.1–4 Thus the designing of new and more efficient anticancer drugs to be used as an alternative to platinum drugs is of immense importance. Among the metal-based new drugs that are currently being developed, ruthenium (Ru)-based drugs attract great attention for their low toxicity and other remarkable properties.2,3,5–9 Two of the Ru(III)-compounds, NAMI-A and KP1019, whose chemical structures are displayed in Fig. 1, have successfully completed phase I and preliminary phase II clinical trials.1,10,11 NAMI-A is mainly used as a targeted antimetastatic drug whereas KP1019 is used as an anticancer agent against primary tumours and metastases and in particular, colon carcinomas.7,12 Besides NAMI-A and KP1019, a large number of Ru-compounds are also being prepared and tested for antitumour activity in cultured tumour cells and animal models.7 The mechanisms of action of Ru-based drugs are considered to be different from those of their platinum counterparts.1,3 To date, two mechanisms have been proposed for NAMI-A and KP1019 drugs: activation-by-reduction and aquation.13–19 However, despite a number of experimental and theoretical investigations, the molecular mechanism of action of NAMI-A, KP1019 and other Ru-based drugs, particularly their mode of interactions and reactions with DNA and other biomolecules, are largely not understood and of much interest.1–3,20,21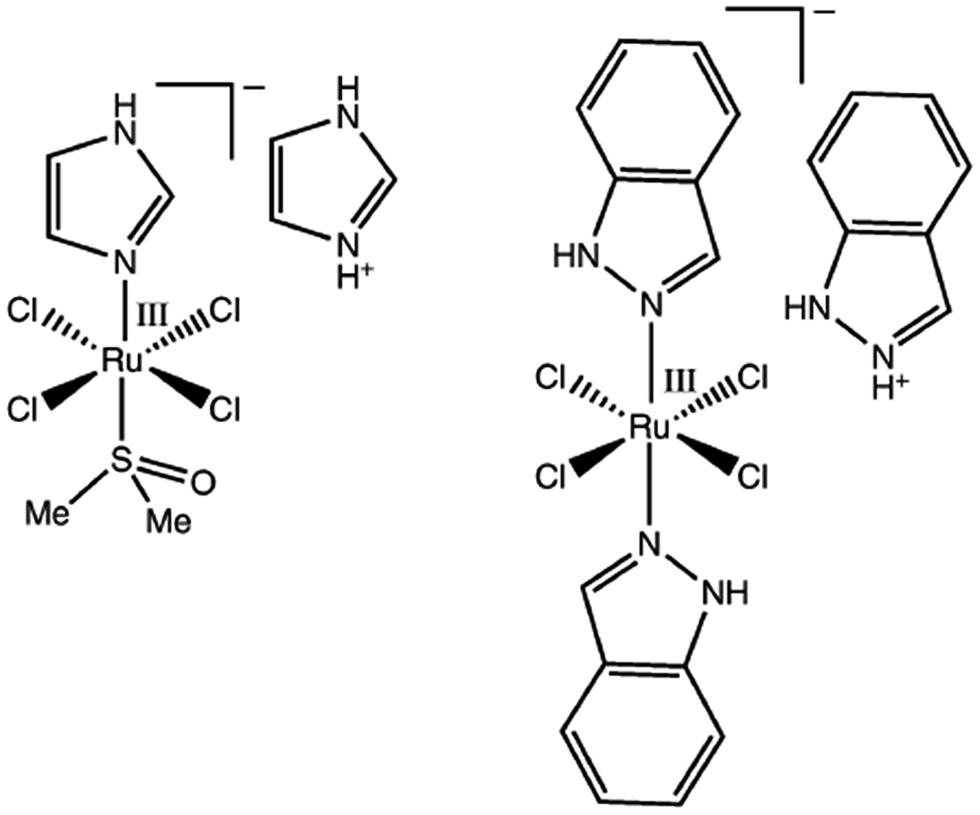 | ||
| Fig. 1 Molecular structures of ruthenium(III) anticancer drugs: (a) NAMI-A and (b) KP1019. | ||
The cytotoxic activities of Ru-based anticancer drugs including NAMI-A and KP1019 may be attributed to their interactions with DNA nucleobases.21–27 It is reported that Ru-based drugs generally bind to the N7 site of guanine but they can also bind to the adenine and cytosine bases of DNA.23,24,28 Groessl et al.28 studied the binding of the Ru-compounds KP1019, NAMI-A and RAPTA-T towards different double-stranded oligonucleotides using electrospray ionisation mass spectrometry and compared it with that of the widely used platinum drugs cisplatin, carboplatin and oxaliplatin. They found that the extent of adduct formation decreased in the following order: cisplatin > oxaliplatin > NAMI-A > RAPTA-T > carboplatin > KP1019.28 A strong preference of organometallic drugs binding to guanine residues was also found in this study.28 Besides DNA, the NAMI-A metabolites were reported to bind to several proteins such as integrins, transferrins and human serum albumin.3,29–31
The metallo(pro)drugs that hydrolyse rapidly are considered to be very cytotoxic.3 It is thought that the Ru-based drugs are hydrolysed readily in vivo to form a number of aquated complexes that are much more reactive toward DNA bases than their parent complexes.3,20 NAMI-A can undergo a hydrolysis reaction in which up to two chloride ligands are substituted by water molecules forming more reactive aquated complexes.13,32,33 However, the quantitative information about the comparative reactivities of NAMI-A and its aquated complexes toward biological targets including DNA bases is not available in the literature. The first hydrolysis step of NAMI-A is reported to be faster than its second hydrolysis step.32,34 The hydrolysis product of NAMI-A has been observed binding to N7 of 9-methyladenine by NMR spectroscopy.13 The hydrolysis mechanisms of Ru-anticancer drugs such as NAMI-A and ICR were investigated thoroughly using density functional theory (DFT).34–37 The bindings/interactions of some of the Ru-complexes with DNA models such as guanine, adenine, base pairs etc. were also investigated theoretically using DFT.38–42 However, to the best of our knowledge, the mechanistic details including barrier energies and rate constants of the reactions of NAMI-A or its aquated complexes with DNA bases are not available in the literature. A thorough study on mechanisms of reactions of NAMI-A and its aquated complexes with DNA bases may be useful in understanding their comparative reactivities and drug's mechanisms of action, and thus may help in rational designing of more efficient new anticancer drugs.
In view of above discussion, we have investigated here theoretically the reactions of NAMI-A, its mono- and di-aquated products (in which one and two chloride ion ligands are replaced by one and two water molecules respectively) at the N7 site of guanine using DFT to understand how aquation/hydrolysis affects the reactivity of NAMI-A towards biological target. As the first hydrolysis step of NAMI-A is reported to be faster than its second hydrolysis step,32,34 the reaction of its mono-aquated product at the N7 site of adenine has also been investigated to explore the preference of drug among the purine bases. The N7 sites of purine bases have been considered in the present study in view of the fact that drug preferably binds to these sites.13,23,24,28
2. Computational details
The calculations in the present contribution were carried out by considering the Ru(III)-anticancer NAMI-A drug, as the [trans-RuCl4(dmso-S)(Im)]− anion, its mono-aquated product in which one chloride anion (Cl−) ligand was replaced by one water molecule, hereafter called NAMI-A (mono-aquated), as the [trans-RuCl3(H2O)(dmso-S)(Im)] neutral and its di-aquated product in which two chloride anion (Cl−) ligands were replaced by two water molecules, hereafter called NAMI-A (di-aquated), as the [trans-RuCl2(H2O)2(dmso-S)(Im)]+cation (Fig. 2) and using the Gaussian09 software suite.43 The structures and vibrational modes were visualized using the GaussView programme.44 The ground state molecular geometries of NAMI-A, NAMI-A (mono-aquated), NAMI-A (di-aquated), guanine, adenine as well as those of the reactant complexes (RCs), transition states (TSs) and product complexes (PCs) involved in different reactions investigated here were fully optimized in gas phase using the Truhlar's M06-2X functional of DFT45 and the (LanL2DZ+6-31G**) hybrid basis set, that is, the effective core potential basis set LanL2DZ for Ru-atom and the standard 6-31G** basis set for all other atoms. These optimization calculations were followed by frequency calculations at the same level of theory, i.e. the M06-2X/(LanL2DZ + 6-31G**) in gas phase, to ensure that each total energy minimum had all vibrational frequencies real, and each TS had only one vibrational frequency imaginary as well as to obtain thermal corrections to enthalpies and Gibbs free energies at 298.15 K. The inputs for TSs involved in the reactions of guanine/adenine were generated broadly following the TS of reaction of first hydrolysis step of NAMI-A, e.g., in case of reaction of NAMI-A at the N7 site of guanine, the O(water) atom in the TS of first hydrolysis step was replaced by the N7 site of guanine and positions/orientations of some atoms/groups of NAMI-A were modified following chemical intuition for generating the input structure for concerned TS. The genuineness of the TSs was confirmed by visually examining the vibrational modes corresponding to their imaginary frequencies. Nonetheless, intrinsic reaction coordinate (IRC) calculations46 were also performed to corroborate the genuineness of TSs. Single point energy (SPE) calculations for all optimized species were carried out using the M06-2X functional and the larger basis set (LanL2DZ+6-311+G**), that is, the effective core potential basis set LanL2DZ for Ru-atom and the 6-311+G** basis set for all other atoms in gas phase and in aqueous solution. The solvent effect of aqueous media was estimated using the conductor-like polarisable continuum model (CPCM).47,48 The thermal corrections to enthalpies and Gibbs free energies at 298.15 K determined using basis set (LanL2DZ+6-31G**) were also applied to the total energies obtained by SPE calculations to obtain the enthalpies and Gibbs free energies of different species in both gas phase and aqueous solution. The rate constant (k) of a reaction was calculated at T = 298.15 K using the following formula:where, ΔG = Gibbs free barrier energy and other symbols have usual meanings.

 | ||
| Fig. 2 Optimized structures of (a) NAMI-A, [trans-RuCl4(dmso-S)(Im)]− anion, (b) NAMI-A (mono-aquated), [trans-RuCl3(H2O)(dmso-S)(Im)] and (c) NAMI-A (di-aquated), [trans-RuCl2(H2O)2(dmso-S)(Im)]+ cation, along with certain interatomic distances (Å) optimized at the M06-2X/(LanL2DZ + 6-31G**) level of theory in gas phase. | ||
It may be noted that the most popular B3LYP functional of DFT49,50 in combination with hybrid basis set (LanL2DZ+6-31G(d,p))/(LanL2DZ+6-31G(d)) had been mostly used in previous studies involving Ru-based drugs.34–39 However, since B3LYP functional is known to underestimate the barrier heights in many cases51 and M06-2X functional is, in general, reported to be a better functional for calculation of barrier heights as well as for thermochemistry of organometallic compounds,51–53 the calculations in this study have been carried out using the M06-2X functional. Nonetheless, in order to check the reliability of the energetics obtained at the M06-2X/(LanL2DZ+6-311+G**)//M06-2X/(LanL2DZ+6-31G**) level of theory in the present study, the barrier energy and rate constant of the first hydrolysis step of NAMI-A where one chloride (Cl−) anion ligand was replaced by a water molecule were calculated at the M06-2X/(LanL2DZ+6-311+G**)//M06-2X/(LanL2DZ+6-31G**) level of theory in gas phase and aqueous media. The mechanism of this reaction is not discussed here because this reaction has already been explored previously34–37 and also this is not our objective here. However, the optimized structures of RC, TS and PC involved in this reaction are presented in Fig. S1 (ESI†) for the sake of completeness of data. The calculated Gibbs free barrier energy (kcal mol−1) and rate constant (s−1) of the first hydrolysis step of NAMI-A are found to be 28.88 (24.01) and 4.06 × 10−9 (1.51 × 10−5), respectively in gas phase (aqueous media) at the M06-2X/(LanL2DZ+6-311+G**)//M06-2X/(LanL2DZ+6-31G**) level of theory (Table S1†). Chen et al.36 reported the rate constant for the same reaction determined at the B3LYP/(LanL2DZ+6-311++G(3df, 2pd))//B3LYP/(LanL2DZ+6-31G(d)) level of theory to be 1.17 × 10−10 s−1 (6.11 × 10−5 s−1) in gas phase (aqueous media). Chen et al.36 also used the CPCM model for solvation calculations. The experimental values of the rate constant of the first hydrolysis step of NAMI-A are reported to be 2.77 × 10−8 to 1.44 × 10−5 s−1 in different buffer concentration (pH 5–8) and 1.44 × 10−5 s−1 in phosphate buffer (pH 7.4) exposed to light.14,54 It is noted that the rate constant of the first hydrolysis step of NAMI-A determined using M06-2X functional is more close to experimental values and is, thus, marginally better than that determined using B3LYP functional. In the present contribution, we would use the geometrical parameters obtained at the M06-2X/(LanL2DZ+6-31G**) level of theory in gas phase and the energetics and rate constants obtained at the M06-2X/(LanL2DZ+6-311+G**)//M06-2X/(LanL2DZ+6-31G**) level of theory in gas phase and aqueous media for discussion of the reactions studied here.
3. Results and discussion
3.1. Reactions at the N7 site of guanine
| Atom | Net charge | ||
|---|---|---|---|
| RC | TS | PC | |
| (a) Reaction of NAMI-A with guanine | |||
| Ru | −0.112 | 0.081 | −0.011 |
| Cl1 | −0.425 | −0.816 | −0.818 |
| Cl2 | −0.432 | −0.358 | −0.439 |
| Cl3 | −0.413 | −0.421 | −0.412 |
| Cl4 | −0.434 | −0.494 | −0.403 |
| S | 1.472 | 1.448 | 1.466 |
| N1 | −0.446 | −0.446 | −0.442 |
| N7 | −0.515 | −0.479 | −0.450 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| (b) Reaction of NAMI-A (mono-aquated) with guanine | |||
| Ru | 0.023 | 0.169 | −0.016 |
| Cl1 | −0.387 | −0.431 | −0.366 |
| Cl2 | −0.369 | −0.302 | −0.401 |
| Cl3 | −0.410 | −0.495 | −0.439 |
| S | 1.470 | 1.407 | 1.458 |
| N1 | −0.448 | −0.439 | −0.439 |
| N7 | −0.556 | −0.519 | −0.447 |
| O(water) | −0.867 | −0.899 | −0.980 |
|
|||
| (c) Reaction of NAMI-A (di-aquated) with guanine | |||
| Ru | 0.264 | 0.263 | 0.174 |
| Cl1 | −0.359 | −0.334 | −0.346 |
| Cl2 | −0.381 | −0.459 | −0.394 |
| S | 1.436 | 1.457 | 1.456 |
| N1 | −0.469 | −0.485 | −0.474 |
| N7 | −0.554 | −0.484 | −0.433 |
| O1(water1) | −0.835 | −0.873 | −0.972 |
| O2(water2) | −0.829 | −0.802 | −0.840 |
|
|||
| (d) Reaction of NAMI-A (mono-aquated) with adenine | |||
| Ru | 0.014 | 0.121 | −0.031 |
| Cl1 | −0.382 | −0.403 | −0.365 |
| Cl2 | −0.348 | −0.244 | −0.375 |
| Cl3 | −0.414 | −0.454 | −0.432 |
| S | 1.478 | 1.443 | 1.465 |
| N1 | −0.456 | −0.475 | −0.455 |
| N7 | −0.566 | −0.577 | −0.478 |
| O(water) | −0.896 | −0.974 | −0.977 |
 | ||
| Fig. 3 Mechanism of reaction of NAMI-A at the N7 site of guanine. Structures of reactant complex (RC1), transition state (TS1) and product complex (PC1) along with certain interatomic distances (Å) obtained at the M06-2X/(LanL2DZ+6-31G(d,p)) level of theory as well as their relative Gibbs free energies (kcal mol−1) obtained at the M06-2X/LanL2DZ+6-311+G(d,p)//M06-2X/(LanL2DZ+6-311G(d,p)) in gas phase (aqueous media) are displayed. | ||
In case of reactions of NAMI-A (mono-aquated) with guanine, it is noted that in the RC2, the Ru–O1(water) bond length is ∼2.15 Å whereas the Ru–Cl bond lengths are found in the range ∼2.37–2.41 Å (Fig. 4). It indicates that water is more tightly attached to the Ru-atom than Cl-ligands in RC2. In going from RC2 to TS2, the Ru–O1(water) bond distance increases to ∼2.6 Å showing water molecule moves away from the Ru-atom whereas the N7(G) moves closer to the Ru-atom of NAMI-A, Ru–N7(G) distance in TS2 decreases to ∼2.69 Å, relative to its value ∼3.93 Å in RC2 (Fig. 4). In going from RC2 to TS2, the other atoms near to Ru-atom are also somewhat displaced, the maximum change occurs in Ru–S bond as it is increased to ∼2.56 Å, relative to its value ∼2.40 Å in RC2 (Fig. 4). In PC2, the O1(water) atom is completely replaced by the N7(G) atom, the Ru–O1(water) and Ru–N7(G) distances being ∼4.19 and 2.14 Å, respectively (Fig. 4). Thus, it is found that the most affected atoms in the reaction of NAMI-A (mono-aquated) at the N7 site of guanine are the Ru, O1(water) and the N7(G). In going from RC2 to PC2, the net charges on Ru and O1(water) are decreased to −0.016 and −0.980|e| respectively, relative to their values 0.023 and −0.867|e| in RC2 (Table 1), while the net charge on N7(G) increased to −0.447|e|, relative to its value −0.556|e| in RC2 (Table 1).
 | ||
| Fig. 4 Mechanism of reaction of NAMI-A (mono-aquated) at the N7 site of guanine. Structures of reactant complex (RC2), transition state (TS2) and product complex (PC2) along with certain interatomic distances (Å) obtained at the M06-2X/(LanL2DZ+6-31G(d,p)) level of theory as well as their relative Gibbs free energies (kcal mol−1) obtained at the M06-2X/LanL2DZ+6-311+G(d,p)//M06-2X/(LanL2DZ+6-311G(d,p)) in gas phase (aqueous media) are displayed. | ||
Similar to the RC2 in reactant complex RC3 of reaction of NAMI-A (di-aquated) also, the water molecules are found to be more tightly attached to the Ru-atom than Cl-ligands, the Ru–O1(water1), Ru–O2(water2) and Ru–Cl1/Cl2 bond distances being ∼2.05, 2.18 and 2.37 Å, respectively (Fig. 5). In going from RC3 to TS3 to PC3, the Ru, O1(water1) and N7(G) atoms are affected much; the O1(water) is completely replaced by the N7(G) and the O1(water1) makes a hydrogen bond with the N9 site of guanine, the hydrogen bonding distance being 1.76 Å (Fig. 5). At the TS3, the bond-breaking (Ru–O1(water1)) and bond-forming (Ru–N7(G)) distances are found to be ∼2.39 and 2.62 Å, respectively (Fig. 5).
 | ||
| Fig. 5 Mechanism of reaction of NAMI-A (di-aquated) at the N7 site of guanine. Structures of reactant complex (RC3), transition state (TS3) and product complex (PC3) along with certain interatomic distances (Å) obtained at the M06-2X/(LanL2DZ+6-31G(d,p)) level of theory as well as their relative Gibbs free energies (kcal mol−1) obtained at the M06-2X/LanL2DZ+6-311+G(d,p)//M06-2X/(LanL2DZ+6-31G(d,p)) in gas phase (aqueous media) are displayed. | ||
| Gas phase | Aqueous media | |||
|---|---|---|---|---|
| ΔH | ΔG | ΔH | ΔG | |
| (a) Reaction of NAMI-A with guanine | ||||
| RC1 | 0.0 | 0.0 | 0.0 | 0.0 |
| TS1 | 42.55 | 44.14 | 26.83 | 28.41 |
| PC1 | 1.64 | 2.04 | −7.21 | −6.81 |
|
||||
| (b) Reaction of NAMI-A (mono-aquated) with guanine | ||||
| RC2 | 0.0 | 0.0 | 0.0 | 0.0 |
| TS2 | 26.31 | 27.14 | 26.31 | 27.15 |
| PC2 | −2.41 | −1.58 | −3.18 | −2.34 |
|
||||
| (c) Reaction of NAMI-A (di-aquated) with guanine | ||||
| RC3 | 0.0 | 0.0 | 0.0 | 0.0 |
| TS3 | 23.30 | 26.23 | 24.23 | 27.17 |
| PC3 | −5.67 | −6.83 | −5.57 | −6.73 |
|
||||
| (d) Reaction of NAMI-A (mono-aquated) with adenine | ||||
| RC4 | 0.0 | 0.0 | 0.0 | 0.0 |
| TS4 | 19.04 | 20.74 | 20.97 | 22.67 |
| PC4 | −4.63 | −3.83 | −4.88 | −4.08 |
For reaction of NAMI-A (mono-aquated) at the N7 site of guanine, the gas phase barrier enthalpy and Gibbs free barrier energy are found to be 26.31 and 27.14 kcal mol−1, respectively (Table 2). It is evident from Table 2 that this reaction would be exothermic in gas phase as the product complex PC2 is more stable than RC2 by ∼2 kcal mol−1. In aqueous media, it is noted that the barrier enthalpy/Gibbs free barrier energy values do not change relative to their values in gas phase and reaction remains exothermic (Table 2). The negligible effect of solvent (aqueous media) on barrier energy of reaction of NAMI-A (mono-aquated) with guanine may be understood as follows. Both the NAMI-A (mono-aquated) and guanine are neutral molecules. The charge distribution and dipole moments do not change noticeably in going from RC2 to TS2, the dipole moment of RC2 (TS2) being 11.77 (11.61) Debye. This would lead to the similar solute–solvent interactions in RC2 and TS2. Therefore, the solvent effects on RC2 and TS2 would be broadly same and cancelled out with each other while calculating the barrier energy. Table 2 shows that reaction of NAMI-A (di-aquated) with guanine is also exothermic as the product complex PC3 is more stable than RC3 by ∼7 kcal mol−1 in gas phase/aqueous media. The barrier energy of reaction involving NAMI-A (di-aquated) is calculated to be 26.23 (27.17) kcal mol−1 in gas phase (aqueous media) (Table 2). Thus our calculations predict that the reactions of both the mono- and di-aquated products of NAMI-A at the N7 site of guanine would occur in both gas phase and aqueous media.
In order to understand the effect of aquation/hydrolysis on the reactions of NAMI-A with biological targets, the Gibbs free barrier energies and rate constants calculated for the reactions of NAMI-A, NAMI-A (mono-aquated) and NAMI-A (di-aquated) with guanine in gas phase and aqueous media are compared (Tables 2 and 3). It is evident from Table 2 that the gas phase Gibbs free barrier energies of reaction of NAMI-A (mono-aquated) and NAMI-A (di-aquated) are substantially lowered to 27.14 and 26.23 kcal mol−1 respectively, relative to its value 44.14 kcal mol−1 of the reaction of NAMI-A with guanine. In aqueous media also, the barrier energies of reactions of mono- and di-aquated products of NAMI-A are somewhat lowered to ∼27.2 kcal mol−1, relative to its value 28.4 kcal mol−1 of the reaction of NAMI-A (Table 2 and Fig. 3–5). The rate constants of the reactions of NAMI-A, NAMI-A (mono-aquated) and NAMI-A (di-aquated) with guanine in gas phase (aqueous media) are found to be 2.6 × 10−20 (9.0 × 10−9), 7.68 × 10−8 (7.55 × 10−8) and 3.57 × 10−8 (7.3 × 10−8) s−1, respectively (Table 3). Thus, our calculations clearly demonstrate that the aquated products of NAMI-A in which one/two chloride anion (Cl−) ligands are replaced by one/two water molecule are appreciably more reactive than NAMI-A itself toward the guanine. Further, the Gibbs free barrier energy and rate constant of the first hydrolysis step of NAMI-A (where a Cl-ligand is replaced by a water molecule) calculated at the same level of theory in gas phase (aqueous media) were found to be 28.88 (24.01) kcal mol−1 and 4.06 × 10−9 (1.51 × 10−5) (s−1), respectively [Table S1†]. The values of the barrier energies and rate constants demonstrate that the first hydrolysis step reaction of NAMI-A is quite faster than the reaction of NAMI-A with guanine [Tables 3 and S1†]. Further, the first hydrolysis step of NAMI-A is reported to be faster than its second hydrolysis step.32,34 In view of the above discussion, it appears that NAMI-A is first hydrolysed to form mainly a more reactive mono-aquated species i.e. NAMI-A (mono-aquated), which then reacts with the biological target like guanine and adenine. This proves a hypothesis on the mechanisms of action of Ru-based drugs, generally accepted by experimentalists.3,13,20,32,33
| Reaction | Rate constant | |
|---|---|---|
| Gas phase | Aqueous media | |
| NAMI-A + guanine | 2.6 × 10−20 | 9 × 10−9 |
| NAMI-A (mono-aquated) + guanine | 7.68 × 10−8 | 7.55 × 10−8 |
| NAMI-A (di-aquated) + guanine | 3.57 × 10−7 | 7.3 × 10−8 |
| NAMI-A (mono-aquated) + adenine | 3.8 × 10−3 | 1.46 × 10−4 |
3.2. Reaction of NAMI-A (mono-aquated) at the N7 site of adenine
The optimized structures of RC4, TS4 and PC4 involved in the reaction of NAMI-A (mono-aquated) at the N7 site of adenine along with certain interatomic distances from Ru-atom (Å) obtained at the M06-2X/(LanL2DZ+6-31G**) level of theory in gas phase are also displayed in Fig. 6. For more geometrical parameters of RC4, TS4 and PC4, please refer to Table S5 (ESI†). The relative energies of these stationary points are also shown in Fig. 6. Structural changes similar to the reaction of NAMI-A (mono-aquated) with guanine e.g. the replacement of water molecule by the N7 site of adenine [N7(A)] and changes in net charges on key atoms (Ru, O1(water) and N7(A)) are also observed in the reactions of NAMI-A (mono-aquated) with adenine (Fig. 4 and 6, Tables S3 and S5†). The Ru–O1(water) and the Ru–N7(A) bond distances in TS3 (PC3) are found to be ∼2.87 (3.85) and 2.93 (2.15) Å, respectively (Fig. 5). It is found that in going from gas phase to aqueous media, the barrier enthalpy and Gibbs free barrier energy are somewhat increased to 20.97 and 22.67 kcal mol−1 respectively, relative to their gas phase values 19.04 and 20.74 kcal mol−1 (Table 2, Fig. 6). The relative enthalpy/Gibbs free energy of product complex PC4 indicates that reaction is exothermic in both gas phase and aqueous media (Table 2, Fig. 6). This and energetics of the reactions of NAMI-A (mono-aquated) and NAMI-A (di-aquated) with guanine (Table 2) indicate that the aquated products of NAMI-A would form adduct at N7 sites of adenine and guanine in agreement with experimental observations.13,23,24,28 | ||
| Fig. 6 Mechanism of reaction of NAMI-A (mono-aquated) at the N7 site of adenine. Structures of reactant complex (RC4), transition state (TS4) and product complex (PC4) along with certain interatomic distances (Å) obtained at the M06-2X/(LanL2DZ+6-31G(d,p)) level of theory as well as their relative Gibbs free energies (kcal mol−1) obtained at the M06-2X/LanL2DZ+6-311+G(d,p)//M06-2X/(LanL2DZ+6-31G(d,p)) in gas phase (aqueous media) are displayed. | ||
Finally, in order to know the comparative reactivity of NAMI-A (mono-aquated) toward the N7 sites of guanine and adenine, the barrier energies and rate constants of these reactions obtained in gas phase and aqueous media are considered. It is noted that the barrier energy of reaction of NAMI-A (aquated) with adenine in gas phase (aqueous media) is appreciably lowered to 20.74 (22.67) kcal mol−1, relative to its value 27.14 (27.15) kcal mol−1 involved in the reaction of NAMI-A (aquated) with guanine. The rate constants for reactions of NAMI-A (aquated) with adenine and guanine are found to be 1.46 × 10−4 and 7.55 × 10−8 s−1 in aqueous media (Table 3). Thus, our calculations predict that NAMI-A (mono-aquated) would react to the N7 site of adenine more favourably than to the N7 site of guanine.
4. Conclusions
Reactions of NAMI-A and its mono- and di-aquated products at the N7 site of guanine as well as the mechanism of reaction of NAMI-A (mono-aquated) at the N7 site of adenine have been investigated using density functional theory. The calculations provide for all the studied reactions, the detailed structural changes occurring from the reactant complex (RC) to transition state (TS) to the product complex (PC), as obtained at the M06-2X/(LanL2DZ+6-31G**) level of theory in gas phase, and the energetics and rate constants obtained at the M06-2X/(LanL2DZ+6-311+G**)//M06-2X/(LanL2DZ+6-31G**) level of theory in gas phase and aqueous media. It is found that both the mono- and di-aquated products of NAMI-A are more reactive than NAMI-A itself toward the N7 site of guanine. It is further found that the NAMI-A (mono-aquated) reacts with the N7 site of adenine more favourably than with the N7 site of guanine, the barrier energy and rate constant of its reaction with adenine (guanine) being 22.67 (27.15) kcal mol−1 and 1.46 × 10−4 s−1 (7.55 × 10−8) in aqueous media. The present study supports the generally accepted experimental hypothesis that NAMI-A is first hydrolysed to form a more reactive mono-aquated species which then reacts with the biological targets like guanine and adenine. Another hypothesis on the mechanism of action of Ru(III)-based drugs through the activation-by-reduction would be the subject of investigation of our next study.Acknowledgements
The authors are thankful to the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Govt. of India, New Delhi for financial support [Research Grant: Fast Track Project No. SR/FTP/PS-047/2012].References
- A. Bergamo, C. Gaiddon, J. Schellens, J. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- I. Bratsos, S. Jedner, T. Gianferrara and E. Alessio, Chimia, 2007, 61, 692–697 CrossRef CAS.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458–470 RSC.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209–233 CrossRef CAS.
- C. S. Allardyce and P. J. Dyson, Dalton Trans., 2016, 45, 3201–3209 RSC.
- E. S. Antonarakis and A. Emadi, Cancer Chemother. Pharmacol., 2010, 66, 1–9 CrossRef CAS PubMed.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817–7823 RSC.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- J. M. Rademaker-Lakhai, D. van den Bongard, D. Pluim, J. H. Beijnen and J. H. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS PubMed.
- G. Sava, S. Zorzet, C. Turrin, F. Vita, M. Soranzo, G. Zabucchi, M. Cocchietto, A. Bergamo, S. DiGiovine and G. Pezzoni, Clin. Cancer Res., 2003, 9, 1898–1905 CAS.
- M. Bacac, A. C. Hotze, K. van der Schilden, J. G. Haasnoot, S. Pacor, E. Alessio, G. Sava and J. Reedijk, J. Inorg. Biochem., 2004, 98, 402–412 CrossRef CAS PubMed.
- M. Bouma, B. Nuijen, M. T. Jansen, G. Sava, A. Flaibani, A. Bult and J. H. Beijnen, Int. J. Pharm., 2002, 248, 239–246 CrossRef CAS PubMed.
- J. Chatlas, R. Van Eldik and B. Keppler, Inorg. Chim. Acta, 1995, 233, 59–63 CrossRef CAS.
- M. Clarke, S. Bitler, D. Rennert, M. Buchbinder and A. Kelman, J. Inorg. Biochem., 1980, 12, 79–87 CrossRef CAS PubMed.
- D. R. Frasca and M. Clarke, J. Am. Chem. Soc., 1999, 121, 8523–8532 CrossRef CAS.
- M. A. Jakupec, E. Reisner, A. Eichinger, M. Pongratz, V. B. Arion, M. Galanski, C. G. Hartinger and B. K. Keppler, J. Med. Chem., 2005, 48, 2831–2837 CrossRef CAS PubMed.
- G. Mestroni, E. Alessio, G. Sava, S. Pacor, M. Coluccia and A. Boccarelli, Met.–Based Drugs, 1994, 1, 41 CrossRef CAS PubMed.
- B. Cebrián-Losantos, E. Reisner, C. R. Kowol, A. Roller, S. Shova, V. B. Arion and B. K. Keppler, Inorg. Chem., 2008, 47, 6513–6523 CrossRef PubMed.
- A. Egger, V. B. Arion, E. Reisner, B. Cebrián-Losantos, S. Shova, G. Trettenhahn and B. K. Keppler, Inorg. Chem., 2005, 44, 122–132 CrossRef CAS PubMed.
- J. Malina, O. Novakova, B. K. Keppler, E. Alessio and V. Brabec, JBIC, J. Biol. Inorg. Chem., 2001, 6, 435–445 CrossRef CAS PubMed.
- V. Brabec and O. Nováková, Drug Resist. Updates, 2006, 9, 111–122 CrossRef CAS PubMed.
- E. Gallori, C. Vettori, E. Alessio, F. G. Vilchez, R. Vilaplana, P. Orioli, A. Casini and L. Messori, Arch. Biochem. Biophys., 2000, 376, 156–162 CrossRef CAS PubMed.
- A. C. Hotze, M. Bacac, A. H. Velders, B. A. Jansen, H. Kooijman, A. L. Spek, J. G. Haasnoot and J. Reedijk, J. Med. Chem., 2003, 46, 1743–1750 CrossRef CAS PubMed.
- A. C. Hotze, E. P. van der Geer, S. E. Caspers, H. Kooijman, A. L. Spek, J. G. Haasnoot and J. Reedijk, Inorg. Chem., 2004, 43, 4935–4943 CrossRef CAS PubMed.
- A. H. Velders, A. C. Hotze, J. G. Haasnoot and J. Reedijk, Inorg. Chem., 1999, 38, 2762–2763 CrossRef CAS PubMed.
- M. Groessl, Y. O. Tsybin, C. G. Hartinger, B. K. Keppler and P. J. Dyson, JBIC, J. Biol. Inorg. Chem., 2010, 15, 677–688 CrossRef CAS PubMed.
- O. Dömötör, C. G. Hartinger, A. K. Bytzek, T. Kiss, B. K. Keppler and E. A. Enyedy, JBIC, J. Biol. Inorg. Chem., 2013, 18, 9–17 CrossRef PubMed.
- J. Sun, Y. Huang, C. Zheng, Y. Zhou, Y. Liu and J. Liu, Biol. Trace Elem. Res., 2015, 163, 266–274 CrossRef CAS PubMed.
- Y. Zhang, A. Ho, J. Yue, L. Kong, Z. Zhou, X. Wu, F. Yang and H. Liang, Eur. J. Med. Chem., 2014, 86, 449–455 CrossRef CAS PubMed.
- G. Sava, A. Bergamo, S. Zorzet, B. Gava, C. Casarsa, M. Cocchietto, A. Furlani, V. Scarcia, B. Serli and E. Iengo, Eur. J. Cancer, 2002, 38, 427–435 CrossRef CAS PubMed.
- L. Messori, F. Kratz and E. Alessio, Met.–Based Drugs, 1996, 3, 1 CrossRef CAS PubMed.
- N. Bešker, C. Coletti, A. Marrone and N. Re, J. Phys. Chem. B, 2008, 112, 3871–3875 CrossRef PubMed.
- J. Chen, L. Chen, S. Liao, K. Zheng and L. Ji, Dalton Trans., 2007, 3507–3515 RSC.
- J. Chen, L. Chen, S. Liao, K. Zheng and L. Ji, J. Phys. Chem. B, 2007, 111, 7862–7869 CrossRef CAS PubMed.
- A. V. Vargiu, A. Robertazzi, A. Magistrato, P. Ruggerone and P. Carloni, J. Phys. Chem. B, 2008, 112, 4401–4409 CrossRef CAS PubMed.
- J.-C. Chen, L.-M. Chen, L.-C. Xu, K.-C. Zheng and L.-N. Ji, J. Phys. Chem. B, 2008, 112, 9966–9974 CrossRef CAS PubMed.
- N. Bešker, C. Coletti, A. Marrone and N. Re, J. Phys. Chem. B, 2007, 111, 9955–9964 CrossRef PubMed.
- C. Gossens, I. Tavernelli and U. Rothlisberger, J. Phys. Chem. A., 2009, 113, 11888–11897 CrossRef CAS PubMed.
- D. Das and P. Mondal, J. Phys. Chem. B, 2015, 119, 10456–10465 CrossRef CAS PubMed.
- C. Gossens, I. Tavernelli and U. Rothlisberger, J. Chem. Theory Comput., 2007, 3, 1212–1222 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, J. R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- R. Dennington, T. Keith and J. Millam, Semichem Inc., Shawnee Mission, KS, 2009.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. Parr, Phys. Rev. A, 1988, 38, 3098 CrossRef.
- S. F. Sousa, P. A. Fernandes and M. J. Ramos, J. Phys. Chem. A, 2007, 111, 10439–10452 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, J. Chem. Theory Comput., 2016 Search PubMed.
- M. Walker, A. J. Harvey, A. Sen and C. E. Dessent, J. Phys. Chem. A, 2013, 117, 12590–12600 CrossRef CAS PubMed.
- M. Bouma, B. Nuijen, M. T. Jansen, G. Sava, A. Bult and J. H. Beijnen, J. Pharm. Biomed. Anal., 2002, 30, 1287–1296 CrossRef CAS PubMed.
- J. Carpenter and F. Weinhold, J. Mol. Struct.: THEOCHEM, 1988, 169, 41–62 CrossRef.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Gibbs free barrier energy and rate constant of the first hydrolysis of NAMI-A are presented in Table S1. The optimized structures of reactant complex, transition state and product complex involved in the hydrolysis of NAMI-A are presented in Fig. S1. The certain optimized geometrical parameters of stationary points involved in the reactions shown in Fig. 3–6 are presented in Tables S2–S5, respectively. See DOI: 10.1039/c6ra24251k |
| This journal is © The Royal Society of Chemistry 2016 |