
Study of the influence of magnetic dilution over relaxation processes in a Zn/Dy single-ion magnet by correlation between luminescence and magnetism†
Jérôme
Long,  *a
Luis D.
Carlos
b
and
*a
Luis D.
Carlos
b
and
*a
Luis D.
Carlos
b
and
Abstract
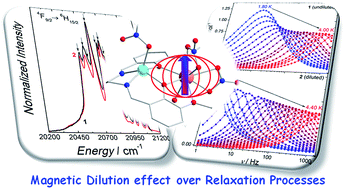
We investigate the magnetic dilution effect on the relaxation mechanisms and the estimation of the energy barrier in a photo-luminescent Dy(III)/Y(III) based Single-Ion Magnet (SIM). The photo-luminescent spectroscopy permits the careful determination of the Orbach barrier, which is in a good agreement with the ab initio calculations. This barrier does not change upon magnetic dilution. The magnetic properties investigations highlight that the determination of the energy barrier is affected by magnetic dilution due to the changes in Quantum Tunnelling of the Magnetization (QTM).
- This article is part of the themed collection: RSC Advances Editors' collection: f Block Chemistry
 Please wait while we load your content...
Please wait while we load your content...

