
Self-cross-linked quaternary phosphonium based anion exchange membranes: assessing the influence of quaternary phosphonium groups on alkaline stability†
Abstract
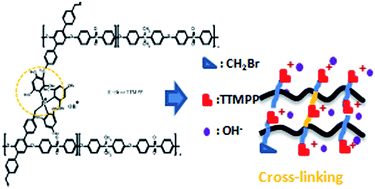
A series of novel quaternary phosphonium functionalized poly(aryl ether sulfone)s were successfully synthesized using a new methyl containing monomer via a multi-step process including bromination, quaternary phosphination and alkalization. The study of the chemical stability of these alkaline anion exchange membranes (AAEMs) using different spectroscopic techniques (1H NMR, 31P NMR and ATR-FTIR) revealed the excellent alkaline stability of the polymer backbone and the partial degradation of phosphonium groups after alkaline exposure at 60 °C for 288 hours. Self-cross-linked membranes with different cross-linking degrees were also synthesized via Friedel–Crafts electrophilic substitution C-alkylation. Despite the fact that the IEC values of the cross-linked membranes are very low, the nano-phase separated morphologies observed by TEM enable the formation of ionic clusters, thus facilitating hydroxide conductivity. Indeed, the normalized IEC hydroxide conductivity for self-cross-linked AAEMs is 13.1 mS g cm−1 meq.−1 at 70 °C, which is comparable to that of other cross-linked AAEMs. The results of ATR-FTIR spectroscopy, TGA, and IEC demonstrated the excellent alkaline stability of the self-cross-linked AAEMs even after 40 days of immersion in 1 M KOH at 60 °C or even under harsher conditions such as 4 M KOH at 80 °C for 16 days, suggesting that the self-cross-linking strategy was beneficial for the protection of phosphonium groups from hydroxide attack.
 Please wait while we load your content...
Please wait while we load your content...

