
Catalyst free decarboxylative trichloromethylation of aldimines†
Eloah P. Ávilaa,
Isabella F. de Souzaa,
Alline V. B. Oliveirab,
Vinicius Kartnallerb,
João Cajaibab,
Rodrigo O. M. A. de Souzab,
Charlane C. Corrêaa and
Giovanni W. Amarante*a
aDepartamento de Química, Universidade Federal de Juiz de Fora, CEP: 36036-900, Juiz de Fora, MG, Brazil. E-mail: giovanni.amarante@ufjf.edu.br
bInstituto de Química, Universidade Federal do Rio de Janeiro, CEP: 22041-909, Rio de Janeiro, RJ, Brazil
First published on 9th November 2016
Abstract
A catalyst free decarboxylative trichloromethylation of imines to afford different trichloromethyl sulfonyl and sulfinyl amines has been presented. Only DMSO as a solvent at room temperature was necessary to provide the corresponding products in good to high isolated yields. A highly diastereoselective version was carried out, leading to the sulfinylimine with good yield and near perfect diastereoselectivity. Regarding the reaction profile, ATR-FTIR spectroscopy was employed to support the entire mechanism and also to provide details on the trichloroacetate salts behavior against electrophiles and in the presence of different solvents.
Introduction
Trichloromethyl groups gained attention due to their applications in the medicinal chemistry and organic synthesis fields.1 The presence of a trichloromethyl group has been pivotal for the biological activity of some molecules, such as Salubrinol, which has been used against Alzheimers as well as for viral infections.2 Furthermore, the trichloromethyl group has also been used for reagents, solvents3 and as precursors in the synthesis of many complex molecules.4 An important synthetic approach is the Jocic reaction, illustrated by Corey and Link in the preparation of chiral-aminoacids, via trichloromethylcarbinol as an intermediate.5 On the other hand, halogenated olefins syntheses were demonstrated by Wahl and co-workers through Michael-type addition of the trichloromethyl anion to E-fumarates.6 These trichloromethyl amine derivatives7,8 are important building blocks in the synthesis of 2,2-dichloro-aziridines.9a,bRegarding the synthesis of trichloromethyl amine derivatives, most likely, these compounds could be accessible through nucleophilic 1,2 addition of trichloromethyl anion to imine derivatives, using trimethyl(trichloromethyl)silane as reagent. However, this reagent has serious drawbacks, such as expensive, restriction over its storage as well as presents low stability.7b
Rozentsveig and co-workers described a protocol to obtain trichloromethyl aryl sulfonamide derivatives.10 The method utilizes seven chemical steps in a one-pot procedure. It took long reaction time and tolerated only aryl sulfonamide groups. Alternative methods to build C–CCl3 bond have been reported in literature.11a,b Lindhardt and co-workers, inspired by Corey,11a reported a protocol to scale-up trichloromethylated alcohols production from addition of trichloromethyl anion to aldehydes using flow12 microreactor conditions.11b This protocol has advantage such as low cost of reagents and most reactions could be carried out at room temperature.
In this work, we present the results of a very simple and effective method for the preparation of trichloromethyl sulfonamides from decarboxylative trichloromethylation of imines. Besides, Attenuated Total Reflection/Fourier Transform Infrared (ATR/FTIR) spectroscopy technique is employed to monitor the reaction components profile. Finally, in order to demonstrate the power of the method, a highly diastereoselective reaction involving a chiral sulfinyl imine is also described.
Results and discussion
Optimization and scope of trichloromethylation decarboxylative reaction
Our studies started with the preparation of sodium and potassium trichloroacetate salts13 and the corresponding N-sulfonyl-aldimines readily accessible following literature protocols.14a,b Having all the substrates in hands, a set of reactions were carried out in order to obtain the best reaction condition to afford trichloromethylated amines (Table 1). The use of aprotic less-polar solvents gave no product (Entries 1 and 2). Switching to DMSO, under microwave irradiation, and catalytic amount of (+/−)-camphorsulfonic acid (CSA),15 the desired product was detected in a 69% of conversion (Entry 3). In order to raise the conversion, CSA was increased at stoichiometric amount, leading to complex mixture of products (Entry 4). This experiment, in fact, showed imine hydrolysis as main product. At this point, in order to avoid the formation of undesired products the temperature was taken to room temperature, combined with three equivalents of salt and without any addition of catalyst or additive. Under this reaction condition, we were able to get the desired product in almost quantitative conversion (Entry 10).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Reaction condition | Solvent | Salt/eq. | Additived | Time | Temperature | Conversiong |
| a Reactions were carried out using 0.5 mmol of 1.b Presence of 0.020 mmol of CSA.c Under microwave irradiation.d One equivalent of CSA.e Using NaTCA.f Presence of 0.010 mmol of CSA.g Based on 1H NMR analysis of the crude reaction mixture. | |||||||
| 1 | Catalytic | PhMe | 2.0 | — | 24 h | 70 | — |
| 2 | Catalyticb | PhF | 2.0 | — | 24 h | 70 | — |
| 3 | Catalyticb,c | DMSO | 2.0 | — | 15 min | 142 | 69 |
| 4 | Non-catalyticc | DMSO | 2.0 | CSA | 15 min | 142 | — |
| 5 | Catalyticb,c | DMSO | 2.0e | — | 15 min | 142 | — |
| 6 | Non-catalytic | DMSO | 2.0 | CSA | 40 min | rt | 98 |
| 7 | Catalyticf | DMSO | 4.0 | — | 40 min | rt | 98 |
| 8 | Catalyticb | DMSO | 4.0 | — | 40 min | rt | 97 |
| 9 | Non-catalytic | DMSO | 4.0 | — | 40 min | rt | 98 |
| 10 | Non-catalytic | DMSO | 3.0 | — | 40 min | rt | 98 |
| 11 | Non-catalytic | DMSO | 2.0 | — | 40 min | rt | 80 |
| 12 | Non-catalytic | DMSO | 1.0 | — | 40 min | rt | 55 |
| 13 | Non-catalytic | DMSO | 3.0e | — | 2 h | rt | — |

Having the standard conditions, various imines were evaluated, considering different electronic and steric characteristics (Table 2). In general, the α-trichloromethyl sulfonamide derivatives were isolated in moderated to good yields (42–87%). For example, electron enriched imine gave the desired product 3h in 85% isolated yield. The presence of a strong electron withdrawing group was also well tolerated. For example, under the optimized reaction condition the corresponding α-trichloromethyl sulfonamide derivative 3m was isolated in 78%. The use of a α,β-unsaturated imine was also evaluated, leading exclusively to the desired 1,2 addition product 3j in 58%. Despite of its low stability, aliphatic imine was also considered, affording the desired product 3o in 42%.

Besides of classical elemental and structural analyses, the C–CCl3 σ bond formation was also confirmed by single crystal X-ray diffraction of adduct 3i (Fig. 1).
 | ||
| Fig. 1 X-ray crystallographic structure of 3i. | ||
ATR/FTIR real-time monitoring on-line experiments
Trichloroacetate salts have been studied by Starke and Möhlmann.16a The procedure described by these authors basically consists on a measurement of dioxide gas absorbed on ascarite in a detachable U-tube weighing after certain intervals. Despite of the good results, this technique has drawbacks in order to obtain concise data on the reaction profile. As an alternative, the real-time infrared spectra technique could be a complementary tool to obtain information on the reaction kinetics, mechanism as well as to elucidate potential reaction intermediates. In this context, Attenuated Transform Infrared (ATR-FTIR), which is a non-destructive and fast monitoring technique employed such as in academic field as industrial processes.17 For example, in situ monitoring of crystallization process, solubility curve measurement, cooling crystallization.17a–dHerein, a series of experiments were submitted and monitored using real time infrared spectroscopy in order to study the trichloroacetate salts profiles in the presence of different solvents and electrophiles (imine and aldehyde). For the experiments, the probe inserted in solution was able to monitor the reaction by the automated spectra acquisition, at each 15 seconds, enabling the observation of the components and their variation due to changes in concentration.
Decarboxylation of potassium trichloroacetate salt (KTCA) in DMSO
We began our studies evaluating how trichloromethyl anions were formed at the reaction conditions developed by us.16 For this purpose, trichloroacetate salt was added to DMSO at room temperature, in which the infrared signal showed the appearance and increase of its bands. After dissolution of KTCA, the decomposition was observed, represented by the decrease in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O bands of the salt (1695 and 1312 cm−1, respectively). At the same time, it was also observed that the C–Cl band of the salt decreased in intensity, while another C–Cl band was formed, with a different frequency. This may indicate the formation of the trichloromethyl anion in solution, as the product of the decarboxylation reaction. Fig. 2 shows the reaction spectra over time (light blue for the beginning of the reaction and dark blue for the end) and the integrated area of the described bands.
O and C–O bands of the salt (1695 and 1312 cm−1, respectively). At the same time, it was also observed that the C–Cl band of the salt decreased in intensity, while another C–Cl band was formed, with a different frequency. This may indicate the formation of the trichloromethyl anion in solution, as the product of the decarboxylation reaction. Fig. 2 shows the reaction spectra over time (light blue for the beginning of the reaction and dark blue for the end) and the integrated area of the described bands.
 | ||
| Fig. 2 Monitoring of the decarboxylation of KTCA in DMSO (1.0 mol L−1). (a) Change in the reaction spectra due to consumption of the salt for the formation of carbon dioxide and trichloromethyl anion (solvent bands subtracted). (b) Integration of the main bands' areas. | ||
As reaction progressed, the intensity of the salt's bands decreased due to its concentration decrease. In Fig. 2b, it is possible to see that the C–Cl band of the salt goes to zero, indicating that close to all KTCA molecules reacted after 30 minutes. The intensity of the CO and C–O band does not zero, due to baseline change over time.
Under heating (140 °C), decarboxylation was also monitored and appeared to occur almost instantaneously. With the addition of the KTCA salt in solution, gas was released right away, and with the ATR-FTIR equipment, only the C–Cl band of the anion (755 cm−1) was seen in the spectra with a relevant intensity (Fig. 3). However, after a few minutes, this anion band started to decrease, as a precipitate was formed in the medium. Since the ATR-FTIR probe used for these experiments can only measure the spectrum of liquids and gas, the precipitated solid could not be measured and the only compound with varying concentration was the anion. This solid may be related to the formation of dichlorocarbene (CCl2) and chloride in solution, as described in the literature16a,18 which could precipitate potassium chloride. This formation was also observed at low temperature (25 °C), but with a slower kinetics; the C–Cl band of the anion took around 240 minutes until it decreased half of its intensity, while it took around five minutes at higher temperature (Fig. 4).
 | ||
| Fig. 3 Solution spectra acquired immediately after addition of KTCA in DMSO at 25 °C and 140 °C (solvent bands subtracted). | ||
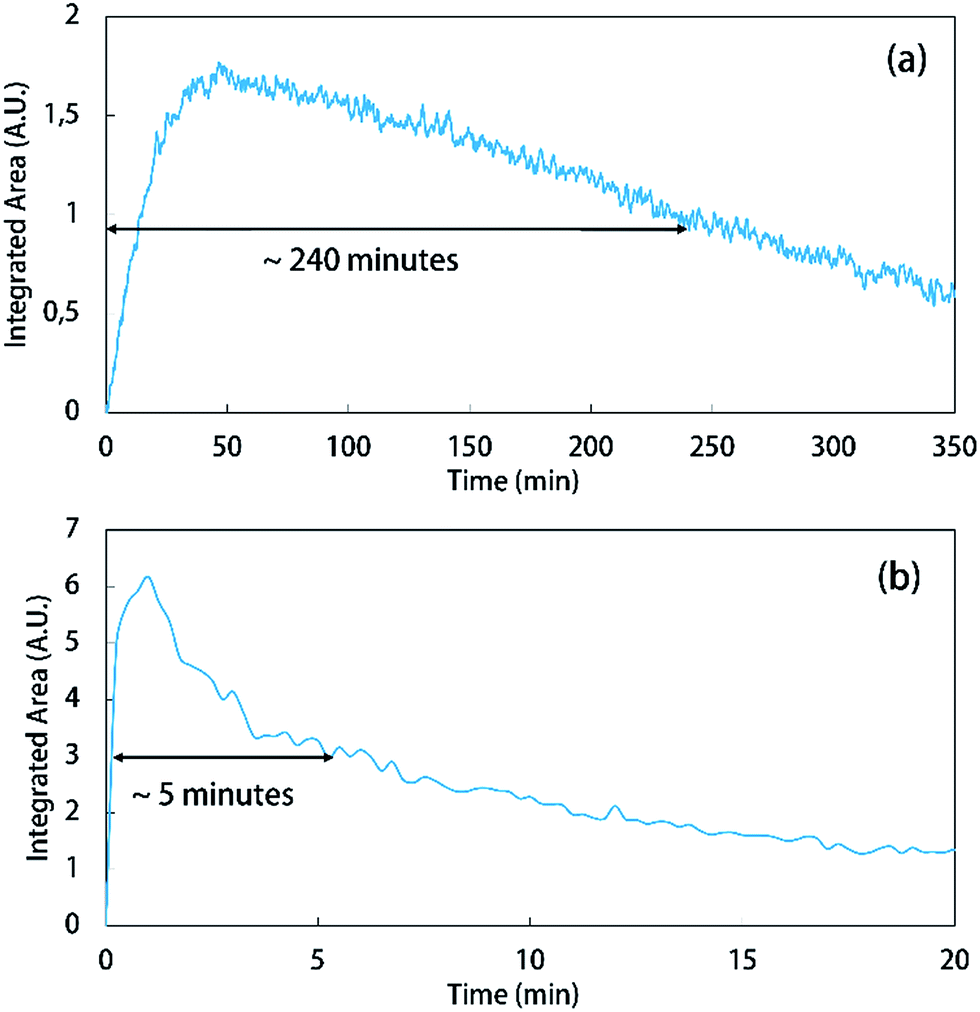 | ||
| Fig. 4 Decrease in the C–Cl band of the formed anion due to product precipitation. (a) At 25 °C; and (b) at 140 °C. | ||
Temperature increase leads to a faster kinetics, both for the decarboxylation reaction and for its possible degradation reaction, to form the carbene and chloride. Hence, for the trichloromethylation reaction, it is important that the decomposition of the trichloromethyl anion does not occur. This might explain why the conversion found for the reactions at higher temperatures was not as high as the ones in room temperature (Table 1).
The decarboxylation of KTCA is described in the literature as a first order kinetics, which means that its concentration varies exponentially as reaction progresses. If the reaction has a rate constant k, the concentration of the salt at a time t can be described as:
| ln[KTCA] = ln[KTCA]o − kt | (1) |
In this case, ln is the natural logarithm, [KTCA] is the concentration at time t and [KTCA]o is the initial concentration of the salt.
Since the data acquired for each decarboxylation reaction is measured over time, an integration approach can be used to estimate the reaction rate constant of this first step, by the linearization of the concentration shown in eqn (1) (plot of the ln[KTCA], measured by the ATR-FTIR, versus time) (Fig. 5).
 | ||
| Fig. 5 Linearization of the salt concentration measured by ATR-FTIR over time for kinetic determination. | ||
Since the linearization could be achieved for experiments with different salt initial concentration, this corroborates the first order mechanism described in the literature. For the plot presented in Fig. 6, the slope of the curves should be close to the same and is related to the reaction rate constant (eqn (1)). For the reactions performed in the conditions presented here, the average constant had a value of 8 × 10−4 s−1.
 | ||
| Fig. 6 Salt in water and ethanol (1.0 mol L−1). (a) C–O band and (b) CO band integration as monitored by ATR-FTIR. | ||
KTCA behavior in protic solvents
The reaction was also performed in protic solvents, such as ethanol and water, but the decarboxylation was not observed, as can be seen in Fig. 6. This behavior is already discussed in the literature, where it is seen that DMSO is indeed a better solvent for decarboxylation reaction, which may be due to stabilization by the solvent, consequently, slower decarboxylation kinetics.16b,cTrichloromethylation of electrophilic species with DMSO
The decarboxylation of KTCA is the first step of the proposed reaction for trichloromethylation. In order for it to occur, it is necessary that trichloromethyl anion is formed in solution, to react with these electrophiles. The complete reaction was also studied using ATR-FTIR, where KTCA was mixed to react with an imine and an aldehyde. First it was evaluated the trichloromethylation of the corresponding imine of molecule 3g (see Table 2). Since the reactional mixture is more complex, few bands are free of interferences from other compound's band. To monitor the course of the reaction a band was chosen related to each molecule: 735 cm−1 for KTCA; 755 cm−1 for CCl3−; 1145 cm−1 for the imine; and 1163 cm−1 for 3g. The latter bands are related to SO bands of the sulfonyl group bonded to the nitrogen. Fig. 7 shows the integrated signal of the bands over time for the experiment where KTCA was added to a solution of the imine in DMSO.
 | ||
| Fig. 7 Monitoring of the trichloromethylation reaction to produce compound 3g, with the addition of the imine the prior to the decarboxylation; integrated area of the bands from each compound, measured by ATR-FTIR. | ||
For this reaction, when KTCA is added in a solution containing the imine, it still needs to decarboxylate in order to generate CCl3− as an intermediate of the reaction. Hence, reaction is controlled by the first step decarboxylation. It can be seen in Fig. 7 that the C–Cl band of the anion increases and the C–Cl band of the KTCA decreases, as was seen in the previous topic. At the same time, the band related to the imine starts to decrease exponentially, while the band related to 3g starts to increase. This happens since the beginning of the decarboxylation, meaning that a little amount of the carbanion in solution is enough to direct the reaction towards the trichloromethylation of the imine. The C–Cl band of the anion does not decrease as it reacts with the imine, because the final product also have a C–Cl band with the same frequency; hence the signal increases by the decarboxylation and is maintained as the anion reacts with the imine.
A factor that should be considered is this first step decarboxylation reaction. In the experiment configuration presented in Fig. 7, the imine was already present in solution as the carbanion was formed to react and form 3g. However, if the carbanion is formed prior to the addition of the imine, the behavior should be different, since the first step would already be over. This experiment was performed and monitored by ATR-FTIR, where KTCA was added to DMSO and let to decarboxylate; after around 40 minutes, the imine was added and reaction was evaluated (Fig. 8).
 | ||
| Fig. 8 Monitoring of the trichloromethylation reaction to produce compound 3g, with the addition of the imine after decarboxylation; integrated area of the bands from each compound, measured by ATR-FTIR. | ||
As can be seen, the prior decarboxylation leads to an almost immediate conversion of the imine to the final compound. The band related to the imine barely changes when it is added to solution, while the band of 3g increases and remains constant throughout the rest of the experiment. This indicates that the rate determining step is indeed the decarboxylation reaction, which is much more slow than the trichloromethylation. This result corroborates what has been discussed in the literature.19 It should be noted that the trichloromethylation was possible after decarboxylation because not much time was given in order that the carbanion could be decomposed in solution.
Diastereoselective application using chiral N-sulfinylimine derivative
The powerful of this method could also be extended to a highly diastereoselective version from a chiral N-tert-butylsulfinyl imine, previously prepared according to the literature procedure.20 After 1.5 h of reaction, at room temperature and with only DMSO as solvent, the chiral α-trichlorosulfinylamine was isolated in 75% yield and with near perfect control of the diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr). It is important to mention the dr was measured by 1H NMR of crude reaction mixture (Scheme 1).
:1 dr). It is important to mention the dr was measured by 1H NMR of crude reaction mixture (Scheme 1).
 | ||
| Scheme 1 Diastereoselective decarboxylative reaction. | ||
Conclusions
In summary, a catalyst free decarboxylative trichloromethylation of imines under mild reaction conditions to afford different trichloromethyl sulfonyl and sulfinyl amines has been presented. Various imines containing either electron-withdrawing, electron-donating and aliphatic groups lead the corresponding adducts in good to high yields and with high chemoselectivity. A highly diastereoselective version was carried out, leading to the sulfinylimine with good yield and near perfect diastereoselectivity.ATR-FTIR spectroscopy were employed to support the entire mechanism and also to provide details on the trichloroacetate salts behavior against electrophiles and in the presence of different solvents.
Experimental section
General procedure and characterization data for the trichloromethyl sulfonamides (3a–o)
To a solution of imine (0.5 mmol) in DMSO (0.7 mmol mL−1) was added potassium trichloroacetate salt (1.5 mmol) and stirred at room temperature for 40 minutes. After completion, it was added dichloromethane (5 mL) and the solution was extracted with water (4 × 10 mL). The aqueous phase was extracted with AcOEt (2 × 20 mL). The volatiles was dried with anhydrous sodium sulfate and evaporated under reduced pressure. The product was obtained after purification through chromatography column (elution: ethyl acetate/hexanes, 3:1).
General procedure and characterization data for the chiral trichloromethyl sulfinimide (5)
To a solution of imine (0.2 mmol) in DMSO d6 (0.7 mmol mL−1) was added potassium trichloroacetate salt (2.0 mmol) and stirred at room temperature for 40 minutes. After completion, it was added dichloromethane (5 mL) and the solution was extracted with water (4 × 10 mL). The aqueous phases was extracted with AcOEt (2 × 20 mL). The volatiles was dried with anhydrous sodium sulfate and evaporated under reduced pressure. The product was obtained after purification through chromatography column (elution: ethyl acetate/hexanes, 3:1)
General procedure of ATR-FTIR monitoring reactions
A ReactIR 45m (Mettler Toledo) spectrometer was used for monitoring potassium trichloroacetate, N-sulfonylimine in DMSO as solvent; it was equipped with an AgX 9.5 mm × 2 m fiber (silver halide), with a 6.35 mm diamond crystal with six internal reflections as an ATR element, ZnSe as a support/focusing element, and a mercury cadmium telluride (MCT) detector using Happ-Genzel apodization. The spectra were acquired in the range of 2000–650 cm−1 with a wavenumber resolution of 8 cm−1 in a 15 s interval between each spectrum (average of 25 scans). All reactions were carried out in a 0.1 L reactor controlled by the EasyMax workstation (Mettler Toledo). The temperature of the reaction was regulated by the equipment with a Pt100 temperature sensor and through a Peltier system jacket. To monitor the reactions, salt (1), and imine and salt (2) solutions were added to the reactor and the temperature was set according to the temperature defined by the design of experiment with a stirring rate of 250 rpm by a mechanical propeller stirrer. The ATR/FTIR probe was immersed in the reaction medium over the course of the whole experiment, and the reaction conversion was evaluated for a stipulated time of 40–120 minutes.Acknowledgements
We would like to thank FAPEMIG (Research Supporting Foundation of the State of Minas Gerais), FAPERJ (Research Supporting Foundation of the State of Rio de Janeiro), CAPES, CNPq and Rede Mineira de Química for financial support.Notes and references
- (a) K. M Borys, M. D. Korzyski and Z. Ochal, Tetrahedron Lett., 2012, 53, 6606–6610 CrossRef; (b) R. D. Baxter and D. G. Blackmond, Tetrahedron, 2013, 69, 5604–5608 CrossRef CAS; (c) A. A. Fesenko, P. P. Solovyev and A. D. Shutalev, Tetrahedron, 2010, 66, 940–946 CrossRef CAS.
- (a) M. Boyce, K. F. Bryant, C. Jousse, K. Long, H. P. Harding, D. Scheuner, R. J. Kaufman, D. Ma, D. M. Coen, D. Ron and J. Yuan, Science, 2005, 307, 935–939 CrossRef CAS PubMed; (b) K. Long, M. Boyce, H. Lin, J. Juan and D. Ma, Bioorg. Med. Chem. Lett., 2005, 15, 3849–3852 CrossRef CAS PubMed.
- F. B. Menezes, H. Gallardo and C. Zucco, Quim. Nova, 2010, 33(10), 2233–2244 CrossRef CAS.
- (a) M. Romero-Ortega, H. Reyes, A. Covarruvias-Zuñiga, R. Cruz and J. G. Avila-Zarragac, Synthesis, 2003, 18, 2765–2767 CrossRef; (b) M. Romero-Ortega, A. Fuentes, C. González, D. Morales and R. Cruz, Synthesis, 1999, 2, 225–227 CrossRef; (c) M. Romero-Ortega, A. Aviles, R. Cruz, Y. Fuentes, R. M. Gomez and A. Plata, J. Org. Chem., 2000, 65, 7244–7247 CrossRef CAS PubMed; (d) S. C Hess, F. Nome, C. Zucco and M. C. Rezende, Synth. Commun., 1989, 19, 3037–3045 CrossRef; (e) J. C. Gesser, C. Zucco and F. Nome, J. Phys. Org. Chem., 1995, 8, 97–102 CrossRef CAS; (f) H. Huo, C. Wang, K. Harms and E. Meggers, J. Am. Chem. Soc., 2015, 137, 9551–9554 CrossRef CAS PubMed; (g) E. Y. Ko, C. H. Lim and K.-H. Chung, Bull. Korean Chem. Soc., 2006, 27, 432–434 CrossRef CAS; (h) V. K. Aggarwal and A. Mereu, J. Org. Chem., 2000, 65, 7211–7212 CrossRef CAS PubMed; (i) N. Wu, B. Wahl, S. Woodward and W. Lewis, Chem.–Eur. J., 2014, 20, 7718–7724 CrossRef CAS PubMed; (j) M. K. Gupta, Z. Li and T. S. Snowden, Org. Lett., 2014, 16, 1602–1605 CrossRef CAS PubMed.
- E. J. Corey and J. O. Link, J. Am. Chem. Soc., 1992, 114, 1906–1908 CrossRef CAS.
- B. Wahl, D. S. Lee and S. Woodward, Eur. J. Org. Chem., 2015, 27, 6033–6039 CrossRef.
- For pharmaceutical applications of trichloromethylamine derivatives, see: (a) Y. Li, Y. Cao, J. Gu, W. Wang, H. Wang, T. Zheng and Z. Sun, Eur. J. Org. Chem., 2011, 4, 676–679 CrossRef; (b) Y. Cao, J. Gu, Y. Li, Z. Sun and H. Wang. PAT-CN101857559, 2013; (c) M. K. Srivastav, N. Srinivasulu and S. M. Shantakumar, Heterocycl. Lett., 2013, 3, 9–16 CAS.
- Related works on the preparation of α-trichloromethylamine derivatives see: (a) M. Therkelsen, M. T. Rasmussen and A. T. Lindhardt, Chem. Commun., 2015, 51, 9651–9654 RSC; (b) B. Wahl, A. Cabré, S. Woodward and W. Lewis, Tetrahedron Lett., 2014, 55, 5829–5831 CrossRef CAS. For related works on synthesis of gamma-amino trichloromethyl compounds, see: (c) H. Morimoto, G. Lu, N. Aoyama, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2007, 129, 9588–9589 CrossRef CAS PubMed; (d) H. Morimoto, S. H. Wiedemann, A. Yamaguchi, S. Harada, Z. Chen, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2006, 118, 3218–3222 CrossRef.
- (a) N. Kimpe, R. Verhé, L. Buyck and N. J. Schamp, J. Org. Chem., 1981, 46, 2079–2081 CrossRef; (b) Y. Li, T. Zheng, W. Wang, Y. Xu, Y. Ma, S. Zhang, H. Wang and Z. Sun, Adv. Synth. Catal., 2012, 354, 308–312 CrossRef CAS.
- Y. A. Aizina, G. G. Levkovskaya and I. B. Rozentsveig, Russ. J. Org. Chem., 2012, 48, 477–480 CrossRef CAS.
- (a) E. J. Corey, J. O. Link and Y. Shao, Tetrahedron Lett., 1992, 33, 3435–3438 CrossRef; (b) A. B. Jensen and A. T. Lindhardt, J. Org. Chem., 2014, 79, 1174–1183 CrossRef CAS PubMed.
- For recent contributions on flow chemistry, see: (a) F. K. Sutili, D. O. Nogueira, S. G. F. Leite, L. S. M. Miranda and R. O. M. A. Souza, RSC Adv., 2015, 47, 37287–37291 RSC; (b) R. A. C. Leão, S. P. Souza, D. O. Nogueira, G. M. A. Silva, M. V. M. Silva, M. L. E. Gutarra, L. S. M. Miranda, A. M. Castro, I. I. Junior and R. O. M. A. Souza, Catal. Sci. Technol., 2016, 6, 4747–4748 RSC; (c) P. F. Carneiro, B. Gutmann, R. O. M. A. de Souza and C. Oliver Kappe, ACS Sustainable Chem. Eng., 2015, 3, 3445–3453 CrossRef CAS; (d) V. D. Pinho, B. Gutmann, L. S. M. Miranda, R. O. M. A. de Souza and C. O. Kappe, J. Org. Chem., 2014, 79, 1555–1562 CrossRef CAS PubMed; (e) I. I. Junior, F. K. Sutili, S. G. F. Leite, K. M. Gonçalves, Y. Cordeiro, I. C. R. Leal, L. S. M. Miranda, M. Ojeda, R. Luque and R. O. M. A. Souza, Green Chem., 2013, 15, 518–524 RSC; (f) B. Pieber and C. O. Kappe, Org. Lett., 2016, 18, 1076–1079 CrossRef CAS PubMed; (g) B. Pieber and C. O. Kappe, Green Chem., 2013, 15, 320–324 RSC.
- For the preparation of potassium trichloroacetate salt: to a solution of trichloroacetic acid (10.5 mmol) in toluene (10 mL) was added potassium hydroxide (10 mmol) and stirred at room temperature for 5 h. The white solid was filtered off under vacuum and washed with cold toluene (2.01 g, quantitative yield).
- For the preparation of aromatic aldimines, see: (a) D. Dong, H. H. Li and S. Tian, J. Am. Chem. Soc., 2010, 132, 5018–5020 CrossRef CAS PubMed; (b) K. Y. Lee, C. G. Lee and N. Kim, Tetrahedron Lett., 2003, 44, 1231–1234 CrossRef CAS For aliphatic aldimines, see: F. Chemla, V. Hebbe and J. Normant, Synthesis, 2000, 1, 75–77 CrossRef.
- For our recent contributions on organocatalysis, see: (a) E. P. Ávila, R. M. S. Justo, V. P. Gonçalves, A. A. Pereira, R. Diniz and G. W. Amarante, J. Org. Chem., 2015, 80, 590–594 CrossRef PubMed; (b) D. L. J. Pinheiro, E. P. Ávila and G. W. Amarante, ChemistrySelect, 2016, 1, 2960–2961 CrossRef CAS; (c) E. P. Ávila, A. C. de Mello, R. Diniz and G. W. Amarante, Eur. J. Org. Chem., 2013, 10, 1881–1883 CrossRef; (d) E. P. Ávila, G. W. Amarante, ChemCatChem, 2012, 4, 1713–1721 Search PubMed; (e) D. L. J. Pinheiro, G. M. F. Batista, J. R. Gonçalves, T. N. Duarte and G. W. Amarante, Eur. J. Org. Chem., 2016, 459–462 CrossRef CAS; (f) A. A. Pereira, P. P. De Castro, A. C. De Mello, B. R. V. Ferreira, M. N. Eberlin and G. W. Amarante, Tetrahedron, 2014, 70, 3271–3275 CrossRef CAS; (g) P. P. De Castro, I. F. Santos and G. W. Amarante, Curr. Org. Synth., 2016, 13, 440–444 CrossRef CAS.
- For studies on decarboxylation of trichloroacetate salts, see: (a) G. Möhlmann and K. Starke, Z. Anorg. Allg. Chem., 1970, 374, 77–85 CrossRef; (b) B. R. Brown and M. A. D. Phil, Q. Rev., Chem. Soc., 1951, 5, 131–146 RSC; (c) W. E. Laque and C. E. Ronneberg, Ohio J. Sci., 1970, 70(2), 97–106 CAS; (d) W. M. Wagner, H. Kloosterziel and A. F. Bickel, Recueil, 1962, 81, 925–932 CrossRef CAS; (e) Z. Pawlak and M. F. Fox, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 1987–1994 RSC.
- For ATR/FTIR applications, see: (a) V. Kartnaller, M. M. V. Romualdo, V. T. V. Lobo and J. Cajaiba, Energy Fuels, 2016, 30, 3660–3666 CrossRef CAS; (b) A. V. A. Souza and J. F. C. Silva, Org. Process Res. Dev., 2012, 17, 127–132 CrossRef; (c) V. Kartnaller, D. C. O. Mariano and J. Cajaiba, Appl. Spectrosc., 2016, 70, 531–538 CrossRef CAS PubMed; (d) V. Kartnaller, I. I. Junior, A. V. A. Souza, I. C. R. Costa, M. J. C. Rezende, J. F. C. Silva and R. O. M. A. Souza, J. Mol. Catal. B: Enzym., 2016, 123, 41–46 CrossRef CAS; (e) M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS; (f) A. V. B. Oliveira, V. Kartnaller, M. S. P. Pedrosa and J. Cajaiba, J. Appl. Polym. Sci., 2015, 132, 42291–42298 Search PubMed; (g) J. Cornel, C. Lindenberg and M. Mazzotti, Ind. Eng. Chem. Res., 2008, 47, 4870–4882 CrossRef CAS; (h) L. R. Knöpke, N. Nemati, A. Köckritz, A. Brückner and U. Bentrup, ChemCatChem, 2010, 2, 273–280 CrossRef; (i) K. Sahre, U. Schulze, T. Hoffmann, M. A. Elrehim, K. J. Eichhorn, D. Pospiech, D. Fischer and B. Voit, J. Appl. Polym. Sci., 2006, 101, 1374–1380 CrossRef CAS; (j) G. Richner, J. A. Bokhoven and Y. Neuhold, Phys. Chem. Chem. Phys., 2011, 13, 12463–12471 RSC. For ATR/FTIR mechanistic studies, see: (k) A. K. Plyamovatyi, R. M. Mukhamadeeva, V. A. Milyukov, O. G. Sinyashin, A. E. Vandyukov, R. R. Shagidullin, A. G. Ginzburg and V. I. Sokolov, Heteroat. Chem., 1995, 6(2), 177–181 CrossRef CAS; (l) G. M. Hamminga, G. Mul and J. A. Moulijn, Chem. Eng. Sci., 2004, 59(22), 5479–5485 CrossRef CAS.
- (a) H. Wynberg, Chem. Rev., 1960, 60(2), 169–184 CrossRef CAS; (b) W. E. Doering, A. K. Hoffmann, J. Am. Chem. Soc., 76(23), 6162–6165 Search PubMed; (c) W. M. Wagner, Proceedings of the Chemical Society, 1959, 229 CAS.
- P. J. Atkins, V. Gold and R. Marsh, J. Chem. Soc., Perkin Trans. 2, 1984, 1239–1245 RSC.
- J. F. Collados, E. Toledano, D. Guijarro and M. Yus, J. Org. Chem., 2012, 77, 5744–5750 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1505456. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra23936f |
| This journal is © The Royal Society of Chemistry 2016 |