
Rhenium(i) complexes with phenanthrolines bearing electron-withdrawing Cl and electron-donating CH3 substituents – synthesis, photophysical, thermal, and electrochemical properties with electroluminescence ability†
Abstract
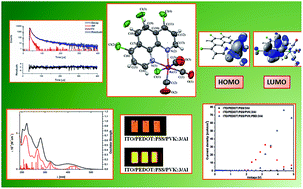
Five rhenium(I) tricarbonyl complexes incorporating 1,10-phenanthroline derivatives with electron-withdrawing Cl and electron-donating CH3 substituents were synthesized, and their photophysical, thermal, and electrochemical properties, with electroluminescence ability were examined. The melting temperature of these complexes was found to decrease from 391 to 281 °C upon adding methyl groups and decreasing the number of chlorine atoms. Compounds bearing methyl substituents could form amorphous molecular materials with glass transition temperatures in the range 118–127 °C. Cyclic voltammetry measurements demonstrated that the complexes are electrochemically active with low energy band gaps in the range 2.24–2.46 eV. All complexes are photoluminescent, form films in the solid state, and blend with poly(9-vinylcarbazole) (PVK) and PVK:(2-(4-tert-butylphenyl)-5-(4-biphenylyl)-1,3,4-oxadiazole) (PBD), emitting light with λem from 560 to 599 nm. They exhibited large Stokes shifts of up to 323 nm in solutions and up to 220 nm as film on a glass substrate. They were tested as guests in organic light-emitting diodes. As hosts, PVK and a binary matrix consisting of PVK with PBD were applied. The ability of the investigated complexes to emit light under an applied voltage was shown.
 Please wait while we load your content...
Please wait while we load your content...

