
Mesoporous template-free gyroid-like nanostructures based on La and Mn co-doped bismuth ferrites with improved photocatalytic activity
Syed Irfanab,
Syed Rizwan*b,
Yang Shena,
Radmila Tomovskacd,
Sonia Zulfiqare,
Muhammad Ilyas Sarwarf and
Ce-Wen Nan*a
aState Key Laboratory for New Ceramics & Fine Processing, School of Materials Science & Engineering, Tsinghua University, Beijing 100084, China. E-mail: cwnan@mail.tsinghua.edu.cn; Fax: +86-10-62771160; Tel: +86-10-62773587
bDepartment of Physics, School of Natural Sciences (SNS), National University of Science & Technology (NUST), Islamabad 44000, Pakistan. E-mail: syedrizwanh83@gmail.com; Tel: +92-51-50855599
cPOLYMAT, University of the Basque Country UPV/EHU, Joxe Mari Korta Center, Avda. Tolosa 72, 20018 Donostia-San Sebastián, Spain
dIkerbasque, Basque Foundation for Science, E-48011, Bilbao, Spain
eDepartment of Chemistry, School of Natural Sciences (SNS), National University of Sciences and Technology (NUST), Islamabad 44000, Pakistan
fDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan
First published on 24th November 2016
Abstract
Template-free and gyroid-like mesoporous nanostructures of La and Mn co-doped bismuth ferrites were fabricated using a simple and low-cost double solvent sol–gel technique. By carefully controlling the amount of precursors and precisely optimizing the calcination process, a well-ordered and crystalline La and Mn co-doped BiFeO3 gyroid-like mesoporous nanostructures network was obtained for photo-degradation applications. The substitution of Mn ions into the Fe sites resulted in a well-ordered mesoporous nanostructure with a flexibility for optical band-gap tuning up to a large extent. Catalytic photo-degradation of Congo red under visible light irradiation occurred at a much higher rate using the mesoporous nanostructures compared to pure BiFeO3 nanoparticles. Thus, large tunability of the band gap, high activity of the mesoporous nanostructure and stability were successfully achieved. The new fabrication method presented here is the first evidence to suggest that such a well-ordered, template-free, gyroid-like mesoporous nanostructure network can be fabricated for low-cost commercial applications.
Introduction
Perovskite bismuth ferrite (BiFeO3) has gained significant attention over the past few years due to its excellent ferroelectric (Curie temperature, Tc ∼ 1100 K) and anti-ferromagnetic (Néel temperature TN ∼ 643 K) properties. This type of multiferroic material exhibits a unique arrangement of magnetic and ferroelectric ordering well above room temperature, which can be modified by an applied electric or magnetic field, respectively.1The coexistence of such diverse characteristics within a single structure makes bismuth ferrite even more striking for applications in light-emitting diodes, magnetic recording media, spintronic devices and ferroelectric solar cells.2–4
Recently, it has been reported that BiFeO3 could be an upcoming photocatalyst because of its visible-light response and low band-gap (2.2–2.7 eV). It also shows good chemical stability5 and an intrinsic electric polarization field, which can allow charge-carrier separation within the semiconducting structure.6
Remarkable photocatalytic activity has been observed with various BiFeO3-based materials; for example, degradation of organic pollutants and oxidation of organic compounds has already been reported.7,8
However, major drawbacks of this exciting photocatalytic activity are poor electron–hole pair separation yield and small surface area. Current efforts have been focused on improving the photocatalytic properties, while growing the structural formulations of BiFeO3 (BFO) at the nanoscale; these consist of zero-dimensional nanoparticles5,9 and one-dimensional nanowires & nanotubes.10,11 More recently, thin film morphologies12 and two-dimensional microplates13 of BFO have shown remarkable activity in the photoelectrochemical oxidation of water for production of oxygen. Nanostructured semiconductors offer a unique aspect to sustained photocatalytic chemistry due to their large accessible pore surface and substantial dimensional reduction of the semiconducting framework. The unique geometrical properties presented by the mesoporous nanostructure and its similar morphologies make certain that the structured materials offer exceptionally fascinating case studies of the complex association between morphologies and optical properties.14–16 Nanostructured bismuth ferrite (BFO) is an important material for a number of applications, such as organic photovoltaics (OPV), environmental remediation and photocatalysis.17–19 For most of these applications, the morphology of the nanostructure determines its performance, which can be adjusted through manipulation of the active surface area.20–22
Moreover, recombination of electron and hole could be effectively suppressed in nanoscale semiconductors due to the short charge carrier diffusion length.23 Therefore, it is sensible to suppose that a high conversion of photon-to-electron yield in these systems. A nanostructured network of BFO with a regular pore structure and a highly exposed surface area holds more potential for improving the photocatalytic efficiency. This is because a 3D open pore structure can provide a large number of active sites, yet can allow a rapid transport of reactants and products through the catalyst.24,25
Herein, for the first time, we adopted a simple and cheap double solvent sol gel method for the fabrication of a well-ordered mesoporous nanostructure of La and Mn doped BFO with highly crystalline pore walls and a large internal surface area. This mesoporous nanostructure exhibits substantially higher photocatalytic activity than that of non-porous BFO under UV-visible illumination. Moreover, the mesoporous nanostructure we obtained consisted of a gyroid-like network, which illustrates the possibility of fabricating self-oriented template-free gyroid networks with improved parameters.
Experimental
Nanoparticles with the general formula Bi1−xLaxFe1−yMnyO3 (abbreviated as BLFMO, x = 0.0, 0.10; y = 0.0, 0.05, 0.15, 0.20), namely Bi0.90La0.10FeO3 (as BLFO), Bi0.90La0.10Fe0.95Mn0.05O3 (as BLFMO-5), Bi0.90La0.10Fe0.85Mn0.15O3 (as BLFMO-15), and Bi0.90La0.10Fe0.80Mn0.20O3 (as BLFMO-20) were synthesized by double solvent sol gel method. Bismuth nitrate pentahydrate (99% pure) and lanthanum nitrate hexahydrate (98.5% pure) were stoichiometrically mixed and dissolved in acetic acid (≥99.5%) and ethylene glycol (≥99.0%), and then stirred for 90 min at 40 °C. Anhydrous iron nitrate (98.5% pure) powder and manganous nitrate (50% sol.) were dissolved in acetic acid with constant magnetic stirring for 90 min at 40 °C. The solutions were then combined and stirred for 180 min at 40 °C. A uniform, reddish brown and fine precursor solution (0.4 M) was produced. To compensate for Bi loss during the heating process, solutions were prepared containing 3% of excess Bi. Ethylene glycol was used as solvent to maintain the electro-negativities of iron & bismuth during the synthesis, whereas acetic acid was used as a catalyst to maintain the solution concentration and for a controlled chemical reaction.26 The as-prepared solution was dried in an oven at 80 °C for 12 h to produce a gel, which was then calcined in a furnace at 600 °C for 3 h. After calcination, the powder was crushed into a fine, homogeneous powder.To verify the structure of the ordered BLFMO samples, we performed wide angle X-ray diffraction measurements. The crystal structure of BLFMO was characterized using a Rigaku 2500 X-ray diffractometer with Cu-Kα radiation source (λ = 0.1542 nm). The scanning angle ranged from 20° and 60° with scanning step of 1° per second. To study the morphology of the BLFMO samples, field-emission scanning electron microscopy (FESEM) was performed (JEOL-7001F) using accelerated voltages of 1.5–3 keV. To avoid the charging effect, samples were sputter-coated with 2–3 nm of platinum before observation. The band-gap and photocatalytic properties of the BLFMO samples were studied using UV-vis diffused reflectance spectra via UV-vis spectrophotometer (Hitachi UV-3310) with an integration sphere. A 300 W xenon lamp with 420 nm filters was used as the visible light source. The distance between the strip lamp and fluid level was kept at 15 cm. The initial concentration of dye solution was 10 mg L−1. The La and Mn co-doped BFO catalyst (100 mg) and an aqueous solution of dye (100 mL) were included in the reaction system. The suspension was magnetically stirred for 120 min in the dark to make sure the mixture had reached adsorption equilibrium before illumination with visible light. During the photoreaction process, 3 mL of the solution was removed after every 30 min interval. The photocatalyst was separated from the solution by centrifugation and the remaining clear liquid was tested by UV visible spectroscopy. The porosity and Brunauer–Emmett–Teller (BET) surface area of BLFMO-5 was obtained using a Quadrasorb-SI v. 5.06 (Quantachrome Instruments Corporation, USA) from N2 sorption/desorption isotherms at 77 K. The BLFMO samples were degassed at 300 °C prior to nitrogen adsorption measurements. The BET surface area was studied using the multi-point method with adsorption calculations in the relative pressure (P/Po) range from 0.05 to 0.25. The pore size distribution was calculated by desorption isotherms using the Barret Joyner Halender (BJH) method.27
Results & discussion
Effect of La and Mn co-doping on crystallization of BFO nanostructure
The calcination condition with slow heating and cooling processes was performed in air using the following procedure: samples were slowly heated from 20 °C to 600 °C at 0.5 °C min−1, held at 600 °C for 3 h, and then slowly cooled from 600 °C to 20 °C at 0.5 °C min−1. Fig. 1 shows the XRD profiles of the BLFMO samples. All diffraction peaks were indexed as rhombohedral with lattice constants a = 5.573 Å and c = 6.915 Å (JCPDS card no. 20-0169), corresponding to (012), (104), (110), (006), (202), (024), (116), (112), (018), and (214) reflections. Compared to diffraction peaks in BFO, BLFO and BLFMO, the (006) and (018) diffraction peaks tended to vanish and (104) and (110) diffraction peaks merged together into a single peak. Moreover, the intensity of the peak related to the (006) plane at around 39°, which arose from the rhombohedral structure, became weaker with the increase in co-substitution.28 The results could be attributed to compositional-driven phase transformation from rhombohedral to orthorhombic. A similar trend was also observed in other reports.29,30 Interestingly, the merged peak of BLFMO, compared to that of the BLFO, shifted toward a lower and then higher angle; a trend which was reversed when the Mn content was 20% or greater; this was inconsistent with other reports.31,32 Negative shift of the merged peak (104) and (110) was observed when 5% of Fe atoms were substituted by Mn atoms. This result may suggest that the rhombohedral distortion was reduced towards the orthorhombic.33 By increasing the Mn concentration, the merged peak position shifted towards higher angle, which may be due to the fact that the lattice constants were decreasing because the ionic radius of Mn2+ (0.645 Å) was relatively lower than that of Fe3+ (0.782 Å).34 The crystallite size of the nanostructures was reduced from BFO to BLFMO-20 (57 nm–20 nm) by increasing concentration of La and Mn into BFO, which was calculated from Scherrer's formula35 after deduction of instrument broadening BS (1°); JADE 5 software was used to fit the values of primitive cell volume. The reduction of grain size and cell volume could be attributed to restricted growth of crystal that results from the substitution element with different radii.36 As can be seen from the enlarged XRD peak between 31 and 33° (Fig. 1), due to a decrease of crystallite size, the inner strain in the crystalline lattice was induced in to BFO, which suggested that XRD peaks broadened as the result of co-doping of La and Mn into BFO nanostructure. Moreover, this lattice strain was due to the nanoscale sized particles, which also have been reported for other metal oxide nanoparticles.37–40 It is important to note that a very small amount of Bi2Fe4O9 impurity was found along with the BFO phase in the co-substituted BLFMO nanostructures. Formation of the impurity phase in the BLFO nanoparticles was suppressed by co-substitution of Mn, which was the main phase of interest for BFO because Bi and/or Fe vacancies led to creation of the impurity phases. Fig. 2a–e shows the FESEM micrographs of pure BFO, BLFO, BLFMO-5, BLFMO-15, and BLFMO-20 nanostructures. It is evident from the figure that BFO and BLFO consisted of non-mesoporous and non-gyroidal nanostructures, lacking pores on the inside of the material. In contrast, clear, well-ordered and gyroid-like mesoporous nanostructure networks were observed for BLFMO-5 and BLFMO-15 samples (Fig. 2c and d), which were devoid of cracks on the surface. It was also clear that the network was continuous to a great extent, which indicated successful fabrication of well-ordered, mesoporous samples without use of a template. From Fig. 2e, it can be seen that the nanostructure network was degraded in BLFMO-20 upon the increment of doping concentration of Mn ≥ 20. | ||
| Fig. 1 1D WAXS profile for BLFMO of different compositions, obtained by La and Mn co-doping at A and B-site of BiFeO3: (a) BFO, (b) BLFO, (c) BLFMO-5, (d) BLFMO-15, and (e) BLFMO-20. | ||
 | ||
| Fig. 2 FESEM micrograph of (a) BFO, (b) BLFO, (c) well-ordered mesoporous nanostructure of BLFMO-5 (the scale bar in the inset is 1 μm), (d) nanostructure of BLFMO-15 featuring less ordered porosity, and (e) crystallized structure of BLFMO-20, in which ordered porosity is not observed. | ||
The pore size distribution and N2 adsorption–desorption isotherms of BLFMO-5 are presented in Fig. 3, demonstrating a typical type-II which may be attributed to the random orientation of pores in the structure and highly crystalline nature of the material.41 Nevertheless, a pore size of 3.5 nm was measured from the differential pore size distribution curve via BJH method (Fig. 3, inset). Table 1 shows the surface area and pore size of all the nanostructures. The narrow hysteresis and pore size distribution of BLFMO-5 clearly indicates the presence of mesopores in the structure.42 Moreover, N2 uptake into micropores was completely absent. Thus, it is deduced that BLFMO-5 is a highly crystalline mesoporous nanostructure.
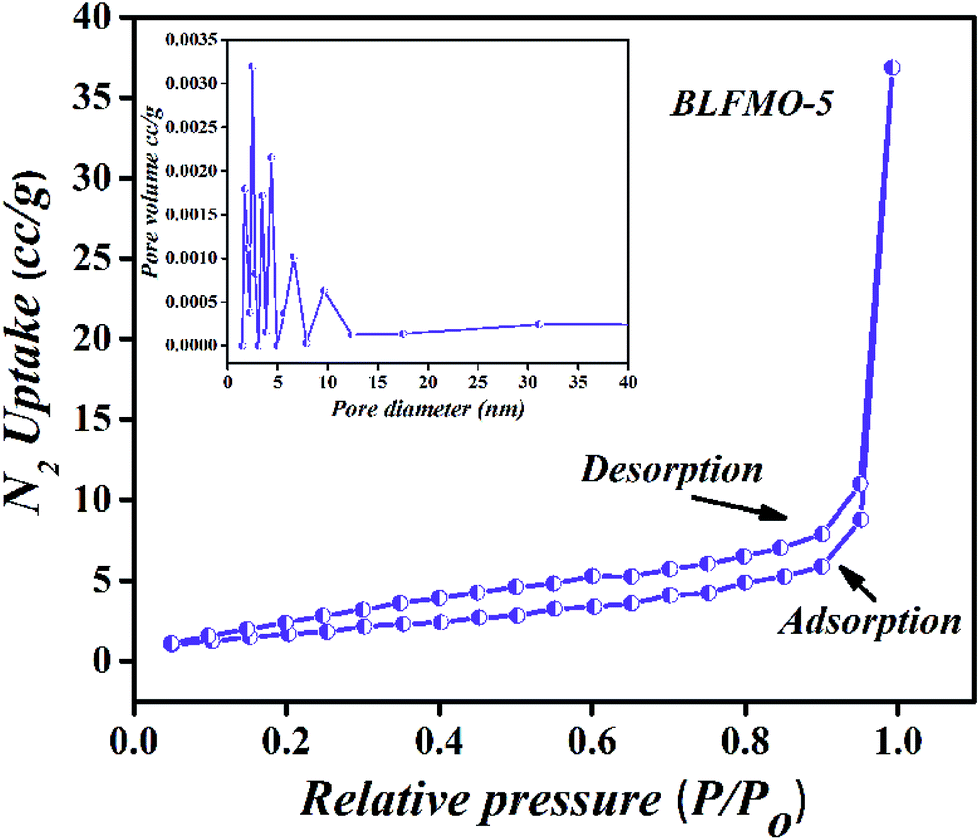 | ||
| Fig. 3 N2 gas isotherms measured at 77 K for BLFMO-5 (inset: differential pore size distribution curves from BJH method). | ||
| Sample | Surface area (m2 g−1) | Pore size (nm) | Pore vol. (cm3 g−1) | Band-gap (eV) | Deg. Eff. (%) |
|---|---|---|---|---|---|
| BFO | 3.3 | 2.2 | 0.02 | 2.08 | 34 |
| BLFO | 5.8 | 2.5 | 0.06 | 2.04 | 70 |
| BLFMO-5 | 9.0 | 3.5 | 0.07 | 1.49 | 97 |
| BLFMO-15 | 6.8 | 2.5 | 0.02 | — | 84 |
| BLFMO-20 | 7.7 | 2.2 | 0.20 | — | 52 |
Band-gap tuning and enhanced photocatalytic performance
It was found that co-doping of La and Mn tuned the optical band-gap of the BFO nanostructures. The diffuse reflectance spectra (DRS) for the band-gap measurement of BLFMO samples are shown in Fig. 4. Two prominent features of the absorption spectra were observed. Firstly, the BLFMO samples exhibited almost the same optical absorption behaviour as for pure BFO nanostructures in the near UV range (300–400 nm), but showed a significantly higher absorption in the visible range (400–800 nm), at the upper limit of our DRS measurements. Secondly, the absorption for BLFMO was much lower in the range of 300–400 nm and overlapped well with BFO and BLFO from 400 nm and beyond. The abrupt drop in absorption beyond 400 nm has also been noticed by others43 and was attributed to the band edge of BFO nanostructures. These features indicated that doping with La and Mn was favorable for optical absorption of BLFMO from visible to IR range. The optical absorption coefficient near the band edge followed the Kubelka–Munk function, (αhν) = A(hν − Eg)n/2 where h, Eg, and ν are Planck's constant, band-gap energy and the light frequency, respectively; ‘A’ is a constant.44 The optical band-gaps of BFO and BLFMO were obtained from the plots of (αhν)2 versus photon energy hν, as shown in the inset of Fig. 4. This relationship provided the band-gap energy (Eg) by extrapolating the straight portion of hν against (αhν)2 plot to a point where α = 0. Accordingly, the band-gap for BFO nanostructure was 2.08 eV, which was quite comparable with the previous results.5,9,45 By increasing the concentration of dopant, the band-gap decreased. The band-gap energy was reduced from 2.08 to 2.04 eV on doping La into BFO, which could be due to lattice strain effect induced by La doping. The reduction of band-gap energy resulted in enhancement of the photocatalytic activity of BFO nanostructure.46 A much smaller band-gap of 1.49 eV was achieved for BLFMO-5, as shown in Table 1. Given the small changes in optical absorption for BLFMO-15 and BLFMO-20 in the visible range, it was quite difficult to calculate the onset of the decrease in optical absorption, as shown in Fig. 4 (inset), which was due to the small amount of impurity phases. The photocatalytic efficiency of the pure and co-doped BFO nanostructures was verified from the degradation of organic pollutant Congo red (CR) under visible light irradiation. Fig. 5a shows the UV-vis absorption spectra of aqueous solution of CR (100 mg L−1) in the presence of BFO nanostructure, which exhibited a strong absorption band in the visible spectral region at 496 nm and a weak one in the UV region at 341 nm. The absorption change at 496 nm was used to follow the degradation of CR, which was examined after every 30 min interval. Fig. 5b and c shows the UV-vis absorption spectra of the CR solution degraded in the presence of BLFMO samples at different irradiation times. The best photocatalytic activity was determined for BLFMO-5, where CR was efficiently decolorized within 2 h. Fig. 5d shows photo-degradation efficiencies as a function of irradiation time under visible light for BFO and BLFMO's. The degradation of CR was nearly negligible under visible light irradiation using pure BFO photocatalyst (blank test). The degradation rate of CR was made significant by introducing BLFMO. On the other hand, the decreased band-gap in the case of nanostructures (demonstrated for BLFMO-5) allowed the formation of larger number of charges upon the same visible light irradiation that induced the oxidation of CR. | ||
| Fig. 4 UV-vis absorption spectra of BFO and BLFMO; the inset is the calculation of their respective bands. | ||
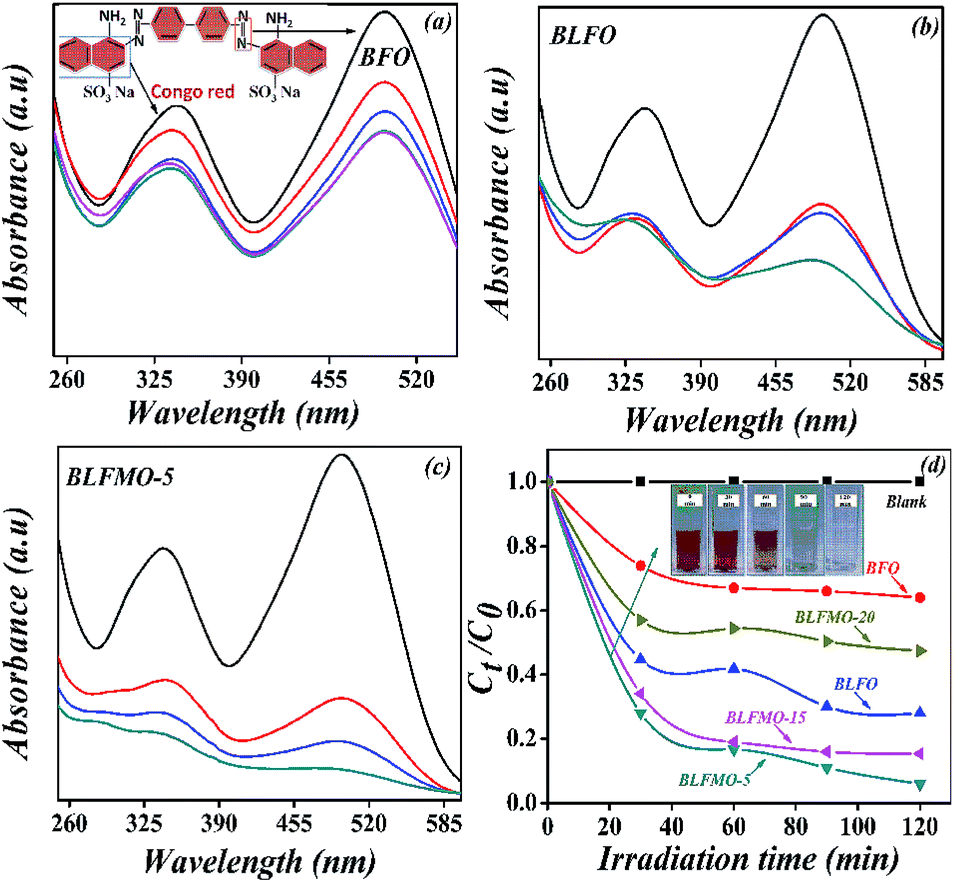 | ||
| Fig. 5 UV-vis absorption spectra of CR solution degraded in the presence of (a) BFO, (b) BLFO, and (c) BLFMO-5. (d) The photo-degradation efficiencies of CR as a function of irradiation time under visible-light for BLFMO and comparison with pure BFO. | ||
 | ||
| Fig. 6 Photoluminescence emission spectra of pure BFO and BLFMO nanostructures (incident light = Xe). | ||
 | ||
| Fig. 7 (a) XRD curve before and after visible-light irradiation (b) stability curves of the photocatalyst BLFMO-5 for degradation of CR under visible light. | ||
Conclusions
In summary, low-cost and simple double solvent sol–gel technique was presented for template-free synthesis of mesoporous gyroid-like nanostructures. By controlling the amount of precursors and precisely optimizing the calcination, well-ordered and crystallized BFO gyroid-like mesoporous nanostructures were obtained with enhanced photocatalytic performance. With the substitution of La and Mn ions onto the Bi and Fe sites, the BLFMO-5 showed a well-ordered gyroid-like mesoporous nanostructure. The band-gap of BFO was successfully tuned to ∼1.49 eV for the BLFMO-5 co-doped sample. Significant enhancement in visible light photocatalytic activity was observed in the BLFMO-5 mesoporous nanostructure; the photo-degradation rate of Congo red under visible light was much higher than that for pure BFO nanostructure, a result that was attributed to its well-ordered porosity and a well-connected nanostructure network. This template-free, gyroid-like nanostructure fabrication approach gives an easy way to fabricate highly porous and well-ordered nanostructured networks for various commercial applications, such as large tunability of the band gap, high photocatalytic activity of nanostructure network and their structural stability. This presents significant advancement in developing commercial-based mesoporous nanostructured materials.Acknowledgements
This study was supported by the National Natural Science Foundation of China (no. 51272121, no. 51221291, no. 51328203 and 51532003). I am thankful to Dr Liangliang Li for useful discussion and for helping me to improve the quality of the manuscript.Notes and references
- G. Catalan and J. F. Scott, Adv. Mater., 2009, 21, 2463–2485 CrossRef CAS; N. Mathur, Nature, 2008, 454, 591–592 CrossRef PubMed.
- T. Choi, S. Lee, Y. J. Choi, V. Kiryukhin and S.-W. Cheong, Science, 2009, 324, 63–66 CrossRef CAS PubMed; A. Tsurumaki, H. Yamada and A. Sawa, Adv. Funct. Mater., 2012, 22, 1040–1047 CrossRef.
- B. Noheda, D. E. Cox, G. Shirane, J. A. Gonzalo, L. E. Cross and S. E. Park, Appl. Phys. Lett., 1999, 74, 2059–2061 CrossRef CAS; R. Guo, L. E. Cross, S. E. Park, B. Noheda, D. E. Cox and G. Shirane, Phys. Rev. Lett., 2000, 84, 5423–5426 CrossRef PubMed.
- H. Bea, M. Gajek, M. Bibes and A. Barthelemy, J. Phys.: Condens. Matter, 2008, 20, 434231–434236 CrossRef CAS; H. T. Yi, T. Choi, S. G. Choi, Y. S. Oh and S.-W. Cheong, Adv. Mater., 2011, 23, 3403–3407 CrossRef PubMed.
- F. Gao, X. Chen, K. Yin, S. Dong, Z. Ren, F. Yuan, T. Yu, Z. Zou and J.-M. Liu, Adv. Mater., 2007, 19, 2889–2892 CrossRef CAS.
- L. Li, P. A. Salvador and G. S. Rohrer, Nanoscale, 2014, 6, 24–42 RSC; Y. Zhang, A. M. Schultz, P. A. Salvador and G. S. Rohrer, J. Mater. Chem., 2011, 21, 4168–4174 RSC.
- K. Rusevova, R. Köferstein, M. Rosell, H. H. Richnow, F.-D. Kopinke and A. Georgi, Chem. Eng. J., 2014, 239, 322–331 CrossRef CAS; S. M. Sun, W. Z. Wang, L. Zhang and M. Shang, J. Phys. Chem. C, 2009, 113, 12826–12831 Search PubMed.
- T. Soltani and M. H. Entezari, J. Mol. Catal. A: Chem., 2013, 377, 197–203 CrossRef CAS; Y. Liu, R. Zuo and S. Qi, J. Mol. Catal. A: Chem., 2013, 376, 1–6 CrossRef; Z. Li, Y. Shen, C. Yang, Y. Lei, Y. Guan, Y. Lin, D. Liu and C.-W. Nan, J. Mater. Chem. A, 2013, 1, 823–829 Search PubMed; Z. Li, Y. Shen, Y. Guan, Y. Hu, Y. Lin and C.-W. Nan, J. Mater. Chem. A, 2014, 2, 1967–1973 Search PubMed; H. Zhao, J. Cao, H. Lv, Y. Wang and G. Zhao, Catal. Commun., 2013, 41, 87–90 CrossRef; M. K. Bhunia, S. K. Das, A. Dutta, A. Sengupta and A. Bhaumik, J. Nanosci. Nanotechnol., 2013, 13, 2557–2565 CrossRef PubMed.
- U. A. Joshi, J. S. Jang, P. H. Borse and J. S. Lee, Appl. Phys. Lett., 2008, 92, 242106–242109 CrossRef.
- X. Zhu, Z. Liu and N. Ming, J. Mater. Chem., 2010, 20, 4015–4030 RSC; S. Mohan and B. Subramanian, RSC Adv., 2013, 3, 23737–23744 RSC.
- S. Li, J. Zhang, M. G. Kibria, Z. Mi, M. Chaker, D. Ma, R. Nechache and F. Rosei, Chem. Commun., 2013, 49, 5856–5858 RSC.
- S. J. A. Moniz, R. Quesada-Cabrera, C. S. Blackman, J. Tang, P. Southern, P. M. Weaver and C. J. Carmalt, J. Mater. Chem. A, 2014, 2, 2922–2927 CAS.
- X. Yang, G. Xu, Z. Ren, X. Wei, C. Chao, S. Gong, G. Shen and G. Han, CrystEngComm, 2014, 16, 4176–4182 RSC.
- H. Y. Hsueh, C. T. Yao and R. M. Ho, Chem. Soc. Rev., 2015, 44, 1974–2018 RSC.
- C. F. Cheng, H. Y. Hsueh, C. H. Lai, C. J. Pan, B. J. H. Wang, C. C. Hu and R. M. Ho, NPG Asia Mater., 2015, 7, 1–8 Search PubMed.
- H. Y. Hsueh, H. Y. Chen, M. S. She, C. K. Chen, R. M. Ho, S. Gwo, H. Haseqawa and E. L. Thomas, Nano Lett., 2010, 10, 4994–5000 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Y. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- S. U. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243–2247 CrossRef CAS PubMed.
- E. Barborini, A. M. Conti, I. Kholmanov, P. Piseri, A. Podesta, P. Milani, C. Cepek, O. Sakho, R. Macovez and M. Sancrotti, Adv. Mater., 2005, 17, 1842–1846 CrossRef CAS.
- D. R. Rolison, Science, 2003, 299, 1698–1701 CrossRef CAS PubMed.
- S. Li, Y. H. Lin, B. P. Zhang, Y. Wang and C. W. Nan, J. Phys. Chem. C, 2010, 114, 2903–2908 CAS.
- L. Fei, J. Yuan, Y. Hu, C. Wu, J. Wang and Y. Wang, Cryst. Growth Des., 2011, 11, 1049–1053 CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- Z. Sun, B. Sun, M. Qiao, J. Wei, Q. Yue, C. Wang, Y. Deng, S. Kaliaguine and D. Zhao, J. Am. Chem. Soc., 2012, 134, 17653–17660 CrossRef CAS PubMed.
- D. Gu, H. Bongard, Y. Deng, D. Feng, Z. Wu, Y. Fang, J. Mao, B. Tu, F. Schüth and D. Zhao, Adv. Mater., 2010, 22, 833–837 CrossRef CAS PubMed.
- Z. W. Chen, J. Q. Hu, Z. Y. X. Lu and H. He, Ceram. Int., 2011, 37, 2359–2364 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- R. Palai, R. S. Katiyar, H. Schmid, P. Tissot, S. J. Clark, J. Robertson, S. A. T. Redfern, G. Catalan and J. F. Scott, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 014110–014121 CrossRef.
- R. Q. Guo, L. Fang, W. Dong, F. G. Zheng and M. R. Shen, J. Phys. Chem. C, 2010, 114, 21390–21396 CAS.
- A. Mukherjee, S. Basu, P. K. Manna, S. M. Yusuf and M. Pal, J. Alloys Compd., 2014, 598, 142–150 CrossRef CAS.
- Z. Wen, G. D. Hu, S. H. Fan, C. H. Yang, W. B. Wu, Y. Zhou, X. M. Chen and S. G. Cui, Thin Solid Films, 2009, 517, 4497–4501 CrossRef CAS.
- S. K. Singh, H. Ishiwara and K. Maruyama, Appl. Phys. Lett., 2006, 88, 262908–262911 CrossRef.
- S. K. Singh, H. Ishiwara, K. Sato and K. Maruyama, J. Appl. Phys., 2007, 102, 094109–094114 CrossRef.
- S. Gupta, A. Sharma, M. Tomar, V. Gupta, M. Pal, R. Guo and A. Bhalla, J. Appl. Phys., 2012, 111, 064110–064116 CrossRef.
- B. D. Cullity, Elements of X-ray diffraction, Addison-Wesleyseries, 2nd edn, 1978 Search PubMed.
- G. S. Arya and N. S. Negi, J. Phys. D: Appl. Phys., 2013, 46, 1–8 CrossRef.
- M. Fukuhara, Phys. Lett. A, 2003, 313, 427–430 CrossRef CAS.
- Z. Wei, T. Xia, J. Ma, W. Feng, J. Dai, Q. Wang and P. Yan, Mater. Charact., 2007, 58, 1019–1024 CrossRef CAS.
- P. M. Diehm, P. Ágoston and K. Albe, ChemPhysChem, 2012, 13, 2443–2454 CrossRef CAS PubMed.
- S. Tsunekawa, K. Ishikawa, Z. Q. Li, Y. Kawazoe and A. Kasuya, Phys. Rev. Lett., 2000, 85, 3440–3443 CrossRef CAS PubMed.
- F. E. Muhammad, N. A. Muhammad and T. He, RSC Adv., 2015, 5, 6186–6194 RSC.
- J. Byun, S. H. Je, H. A. Patel, A. Coskun and C. T. Yavuz, J. Mater. Chem. A, 2014, 2, 12507–12512 CAS.
- N. Miriyala, K. Prashanthi and T. Thundat, Phys. Status Solidi RRL, 2013, 7, 668–671 CrossRef CAS.
- P. Kubelka and F. Munk, Z. Tech. Phys., 1931, 12, 593–601 Search PubMed.
- R. Guo, L. Fang, W. Dong, F. Zheng and M. Shen, J. Mater. Chem. C, 2010, 114, 21390–21396 CAS.
- N. S. A. Satar, A. W. Aziz, M. K. Yaakob, M. Z. A. Yahya, O. H. Hassan, T. I. T. Kudin and N. H. M. Kaus, J. Phys. Chem. C, 2016, 120(45), 26012–26020 CAS.
- A. F. Vladimir, A. A. Khan and A. B. Alexander, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 165317–165325 CrossRef.
- K. Prashanthi, G. Thakur and T. Thundat, Surf. Sci., 2012, 606, L83–L86 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |