
Synthesis and properties of polyurethane urea with pyridine-2,6-dicarboxamide moieties in their structure
Abstract
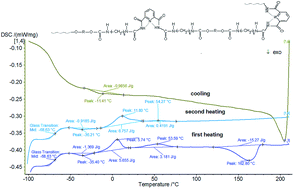
The synthesis of new heterocyclic polyurethane materials is an attractive way to study structure–property relationships. In this work we report on the introduction of 2,6-pyridinedicarboxamide into the main chains of linear and crosslinked polyurethanes. These materials were synthesized using the same soft block (Terathane 1400) and different hard blocks via a two-step polymerization method. The FTIR spectra indicated that the inclusion of pyridine derivative into polyurethane urea chain structure had been successful. The obtained polyurethane matrix structure was characterized through fewer specific intermolecular hydrogen bonds between the pyridine units, urethane and amide groups, due to the fact that the 2,6-pyridinedicarboxamide units are likely to adopt syn–syn conformations when in solid state. These heterocyclic polyurethanes also exhibit improved thermal stability (as measured by TGA) compared with conventional polyurethanes. Stress–strain curves show that the tensile strength and elongation at break are higher in the case of crosslinked samples. The properties of the linear heterocyclic polyurethanes were all compared to the properties of the crosslinked heterocyclic polyurethanes.
 Please wait while we load your content...
Please wait while we load your content...

