
Doping structure and degradation mechanism of polypyrrole–Nafion® composite membrane for vanadium redox flow batteries†
Abstract
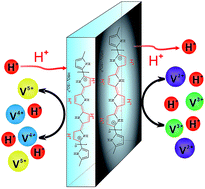
A HPPY–N212 composite membrane was prepared by the in situ polymerization of pyrrole on Nafion® 212 substrate membrane, followed by sulfuric acid treatment. The proton-acid doping structure endowed the HPPY–N212 membrane with enhanced conductivity as well as reduced vanadium ion permeability. These unique properties enabled vanadium redox flow battery (VRFB) fabricated with HPPY–N212 membrane to exhibit better coulombic, voltage and energy efficiency than that with N212 membrane under current densities of 60–150 mA cm−2. However, a gradual decay of voltage and energy efficiency occurred during the charge–discharge cycles. The efficiency decay resulted from the irreversible damage to PPY doping structure caused by the over-oxidation during charge–discharge cycles. These investigations help better understand the structure–performance relationships and open up exciting opportunities for the development of new high-performance membranes for VRFBs.
 Please wait while we load your content...
Please wait while we load your content...

