DOI:
10.1039/C6RA23547F
(Paper)
RSC Adv., 2016,
6, 108335-108342
Synthesis and characterization of a series of novel polyoxometalate-supported carbonyl manganese derivatives†
Received
22nd September 2016
, Accepted 7th November 2016
First published on 8th November 2016
Abstract
Four novel heteropolytungstate-supported carbonyl manganese derivatives [(M4(H2O)10)(XW9O33)2{Mn(CO)3}2]n− (X = Sb/Bi; M = Mn/Mn3.5W0.5) have been successfully synthesized and characterized by single crystal X-ray diffraction crystallography, IR and UV spectroscopy, representing the first examples of structurally characterized transition metal substituted sandwich-type tungsto-antimonates/bismutates incorporated with carbonyl manganese groups. The organic–inorganic hybrids are composed of two {Mn(CO)3} groups attached to a dimeric heteropolytungstate {M4(B-β-XW9O33)2} unit via six MnI–O–W bonds. The {M4(B-β-XW9O33)2} unit is built up of a central symmetric parallelogram-like tetra-MnII cluster sealed into two identical [B-β-XW9O33]9− fragments through MnII–O–W bonds. Furthermore, polyanions 1 and 2 proved to be efficient for the electrocatalytic reduction of NO2−. Magnetic measurements on these complexes were also investigated.
Introduction
Recently, considerable attention has been directed toward polyoxometalate (POM)-based organometallic derivatives because of their versatile structures and fascinating properties, resulting in multiple potential applications in catalysis, electro/photo-chromism, magnetism and medicine.1,2 In this context, immobilization of metal carbonyl units onto the POMs surface are expected to obtain functional compounds and emerge as an important category.3 POM-based metal carbonyl derivatives (PMCDs) possess the dual advantages of metal carbonyl groups and POMs. Therefore, a great deal of research on their extraordinary structures, synthetic strategies, and catalytic properties of these inorganic–organic hybrids have been investigated in detail in the past few years.4–7 However, most of the reported compounds are still based on Lindqvist-type polyoxometalates.4 In comparison, the reports on Keggin-type PMCDs remain relatively rare.5 In the course of our research on PMCDs, we have reported preparation and structural characterization of various heteropolyoxometalate-based metal carbonyl derivatives, {(A-α-H2XW9O34)Mn(CO)3} (X = Ge or Si),5f {(XW11O39)[Re(CO)3]3(OH)2} (X = As, P),5i,j {[PMo3O16][Re(CO)3]4},5k {[Mn4(H2O)10](TeW9O33)2[Mn(CO3)]2}5l and several isopolyoxometalate-supported metal carbonyl derivatives, {Mo6O16(OCH3)2[HOCH2C(CH2O)3]2[Mn(CO)3]2},6a {(M8O30)[M′(CO)3]2} (M = W, Mo; M′ = Re, Mn),6c,d {(W5O18)[(Re(CO)3)4][(OH)(O)]}.4g A comprehensive literature survey of the condensation of Keggin-type PMCDs is shown in Table 1.
Table 1 Summary of Keggin-type PMCDs
| Years |
Formulas |
Precursors |
Ref. |
| 1979 |
[CpFe(CO)2(SnW11PO39)]4−, [(OC)3Co(SnW11SiO39)2]11−, [{CpFe(CO)2Ge}2W11SiO40]4−, and related compounds |
[PW11O39]8−, [SiW11O39]8− |
5a |
| 1983 |
[{CpFe(CO)2Sn}2W10PO38]5− |
Na2WO4/NaH2PO4 |
5b |
| 1990 |
[(Ph3P)2Rh(CO)(CH3CN)]n[XM12O40] (X = P, Si; M = Mo, W; n = 3, 4) |
[XM12O40]4− (X = P, Si; M = Mo, W) |
5c |
| 2008 |
[α-SiW11O39RuII(CO)]6− |
[α-SiW11O39]8− |
5d |
| 2012 |
[Mn(CO)3(CH3CN)3]3[α-XM12O40] (X = P, Si, Ge; M = Mo, W) |
[α-XM12O40] |
5e |
| 2012 |
[(OC)3Mn(A-α-H2XW9O34)]8− (X = Ge/Si) |
[γ-XW10] (X = Ge/Si) |
5f |
| 2013 |
[X2W20O70{M(CO)3}2]12− (M = Mn/Re, X = Sb/Bi) |
[X2W22O74(OH)2]12− (X = Sb/Bi) |
5g |
| 2014 |
[P2W23O80{M(CO)3}2]10− (M = Re/Mn) |
[α-PW11O39]7− |
5h |
| 2014 |
[H11(AsW11O39)4{(Re(CO)3)3(μ3-OH)(μ2-OH)}4]13− |
[HAsW9O34]8− |
5i |
| 2014 |
[(PW11O39){Re(CO)3}3(μ3-O)(μ2-OH)]428− |
Na2WO4·2H2O, 85% H3PO4 |
5j |
| 2015 |
[(PMo3O16){Re(CO)3}4]5− |
(NH4)6Mo7O24·4H2O, Na2HPO4·12H2O |
5k |
| 2015 |
[{Mn(CO3)}(Mn(H2O)2)(Mn(H2O)3)(TeW9O33)]26− |
[TeW9O33]8− |
5l |
On the other hand, lacunary POMs can be regarded as inorganic multidentate ligands, with vacant sites containing terminal and basic oxygens that can coordinate to electrophilic center, particularly transition metal ions, forming the well-known transition-metal-substituted polyoxometalates with magnetic properties and catalytic properties.8 Therefore, we decided to carry out a systematic study on the chemical system based on the manganese carbonyl, transition metal and trivacant polyoxometalates.
Herein, we report on four novel Keggin-type PMCDs (NH4)3H5[{Mn4(H2O)10}(β-BiW9O33)2{Mn(CO)3}2]·31H2O (1) Na6[(CH3)4N]2[{Mn4(H2O)10}(β-SbW9O33)2{Mn(CO)3}2]·36H2O (2), [(CH3)4N]2{Mn(H2O)6}2[{Mn3.5W0.5(H2O)10}(β-SbW9O33)2{Mn(CO)3}2]·12H2O (3), [(CH3)4N]2{Mn(H2O)6}2[{Mn3.5W0.5(H2O)10}(β-BiW9O33)2{Mn(CO)3}2]·12H2O (4). To the best of our knowledge, these complexes are the first examples structurally characterized transition metal substituted sandwich-type tungsto-antimonates/bismutates incorporated with carbonyl manganese groups.
Results and discussion
Synthesis
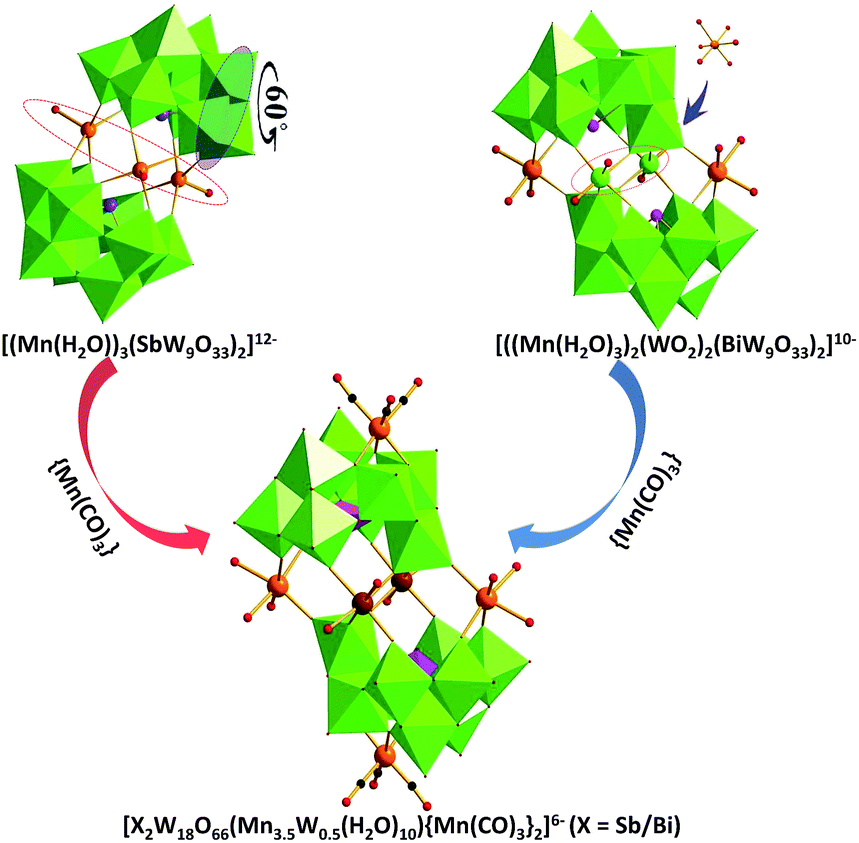
In the past few years, we have prepared and thoroughly characterized some PMCDs by utilizing lacunary POMs as chelating ligands to incorporate metal multicarbonyl units by conventional aqueous solution method. As part of our continuing work, we have been attempting to introduce the transition metal (TM) into PMCDs, leading to products with interesting magnetic and electrochemical properties. Although there are unavoidable competitive reactions among highly negative POM precursors, electrophilic groups {M(CO)3} (M = Mn, Re) and less active TM ions in the same reaction system. TM, a kind of “connectors”, can play an important role in the connection of lacunary POMs fragments.8,9 In this article, tri-lacunary polyoxoanions [XW9O33]9− (X = Sb/Bi) were chosen as precursors to react with [Mn(CO)3]+ and Mn2+ ions. As expected, two new compounds 1 and 2 are successfully prepared in a mixed solvent H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3CN solution (volume ratio, 4–5:1). When the volume ratio of H2O:CH3CN is lower than 4, some oily product was obtained, which has been proved to be Mn(CO)5Br by IR results. However, no crystals can be isolated if the volume ratio is higher than 5. In addition, the reaction is particularly sensitive to the pH value because compounds could be isolated with pH ranging from 5 to 7, with the pH value 6.5 giving the highest yield. The architecture of 1 or 2 can be regarded as two {Mn(CO)3} fragments attached onto a simple sandwich skeleton comprising two [β-XW9O33]9− subunits coordinated to four transition metals. This may indicate that transition metals are easier to occupy the vacant sites of POMs than metal carbonyl fragments. On the other hand, we also utilize the pre-obtained transition metal-substituted POMs [{MnII(H2O)}3(SbW9O33)2]12− (5) and [(MnII(H2O)3)2(WO2)2(BiW9O33)2]10− (6) as precursors to react with {Mn(CO)3} fragments. Interestingly, the sandwich building block is maintained in the final products 3 and 4, supporting our above-mentioned assertion (Scheme 1).
:CH3CN solution (volume ratio, 4–5:1). When the volume ratio of H2O:CH3CN is lower than 4, some oily product was obtained, which has been proved to be Mn(CO)5Br by IR results. However, no crystals can be isolated if the volume ratio is higher than 5. In addition, the reaction is particularly sensitive to the pH value because compounds could be isolated with pH ranging from 5 to 7, with the pH value 6.5 giving the highest yield. The architecture of 1 or 2 can be regarded as two {Mn(CO)3} fragments attached onto a simple sandwich skeleton comprising two [β-XW9O33]9− subunits coordinated to four transition metals. This may indicate that transition metals are easier to occupy the vacant sites of POMs than metal carbonyl fragments. On the other hand, we also utilize the pre-obtained transition metal-substituted POMs [{MnII(H2O)}3(SbW9O33)2]12− (5) and [(MnII(H2O)3)2(WO2)2(BiW9O33)2]10− (6) as precursors to react with {Mn(CO)3} fragments. Interestingly, the sandwich building block is maintained in the final products 3 and 4, supporting our above-mentioned assertion (Scheme 1).
 |
| | Scheme 1 Preparation of novel PMCDs 1, 2, 3 and 4. Colour code: O, red balls; C, black balls; Mn/Mn(W), orange balls; WO6 octahedra, green; XO3 (X = Bi/Sb) tetrahedra, purple. | |
Structure descriptions
All the crystallographic data for compounds 1, 2, 3, and 4 are listed in Table 2. The four corresponding polyanions [{Mn4(H2O)10}(β-BiW9O33)2{Mn(CO)3}2]8− (1a), [{Mn4(H2O)10}(β-SbW9O33)2{Mn(CO)3}2]8− (2a), [{Mn3.5W0.5(H2O)10}(β-SbW9O33)2{Mn(CO)3}2]6− (3a), and [{Mn3.5W0.5(H2O)10}(β-BiW9O33)2{Mn(CO)3}2]6− (4a) have the similar molecular structures. Single-crystal X-ray analyses revealed that all title polyanions consist of two {Mn(CO)3} groups attached to the usual M4X2W18 POM framework (Fig. 1b).
Table 2 Crystal structure data for compounds 1, 2, 3 and 4
| |
1 |
2 |
3 |
4 |
| Formula |
C6H94Bi2Mn6N3O113W18 |
C14H116Mn6N2Na6O118Sb2W18 |
C14H92Mn7.5N2O106Sb2W18.5 |
C14H92Bi2Mn7.5N2O106W18.5 |
| Mr (g mol−1) |
6073.74 |
6221.46 |
6041.67 |
6216.13 |
| T (K) |
296(2) |
296(2) |
293(2) |
293(2) |
| Space group |
C2/m |
P2(1)/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
Triclinic |
| a (Å) |
25.879(16) |
16.4654(12) |
12.5810(19) |
12.663(3) |
| b (Å) |
14.409(8) |
14.4945(10) |
13.282(2) |
13.413(3) |
| c (Å) |
16.235(10) |
25.9004(18) |
19.926(3) |
20.342(5) |
| α (deg) |
90 |
90 |
77.307(2) |
76.453(4) |
| β (deg) |
105.016(10) |
105.3810(10) |
86.633(2) |
85.953(4) |
| γ (deg) |
90 |
90 |
63.697(2) |
63.542(4) |
| V (Å3) |
5847(6) |
5959.9(7) |
2909.1(8) |
3004.9(13) |
| Limiting indices |
−30 ≤ h ≤ 29, −17 ≤k ≤ 11, −19 ≤ l ≤ 19 |
−19 ≤ h ≤ 18, −17 ≤ k ≤ 7, −30 ≤ l ≤ 28 |
−14 ≤ h ≤ 7, −15 ≤ k ≤ 15, −23 ≤ l ≤ 22 |
−14 ≤ h ≤ 15, −15 ≤ k ≤ 15, −24 ≤ l ≤ 24 |
| GOF on F2 |
1.052 |
1.010 |
1.043 |
1.001 |
| R1, wR2 [I > 2σ(I)] |
0.0509, 0.1047 |
0.0333, 0.0719 |
0.0405, 0.1036 |
0.0593, 0.1163 |
| R1, wR2 [all data] |
0.0991, 0.1162 |
0.0500, 0.0761 |
0.0511, 0.1078 |
0.1124, 0.1305 |
 |
| | Fig. 1 (a) Ball-and-stick and polyhedral representations of polyanion 1a; (b) ball-and-stick representations of M4X2W18; (c) the central symmetric parallelogram-like tetra-MnII cluster in 1a; (d) {Mn(CO)3} group in polyanion 1a; (e) {Mn(CO)3} group in the reported {Mo8[Mn(CO)3]2} cluster.6d Colour code: O, red balls; C, black balls; Mn, orange balls; W, green balls; Bi, purple balls; WO6 octahedra, green; BiO3 tetrahedra, purple; MoO6 octahedra, dark green. Cations and water molecules are omitted for clarity. | |
In polyanion 1a, the M4X2W18 unit represents the Mn(II)-analogue of Kortz's Fe(III) and Al(III)-containing polyanions.10 In full analogy, it comprises two [β-BiW9O33]9− subunits joined by a central pair and a peripheral pair of Mn(II) ions, resulting in a structure with idealized C2h symmetry (Fig. 1b). The inner two Mn2+ ions (Mn2, Mn2A) have two terminal H2O ligands and the outer two Mn2+ ions (Mn3, Mn3A) have three terminal H2O ligands. The Mn2 ion is octahedrally coordinated by two atoms from one [β-BiW9O33]9− fragment [Mn–O: 2.083(6), 2.138(4) Å], two oxygen atoms from the other [β-BiW9O33]9− subunit [Mn–O: 2.089(3), 2.134(1) Å] and two O atoms from two terminal H2O molecules [Mn–O: 2.252(0), 2.259(6) Å], whereas the octahedral geometry of Mn3 ion is constituted by two O atoms from one [β-BiW9O33]9− fragment [Mn–O: 2.151(0), 2.166(8) Å], one oxygen atom from the other [β-BiW9O33]9− segment [Mn–O: 2.120(2) Å] and three oxygen atoms from three terminal water molecules [Mn–O: 2.178(1)–2.2077 Å]. Interestingly, the four Mn atoms form a parallelogram with the neighboring two edge distances 5.773 and 5.869 Å, respectively (Fig. 1c).
Most notably, each {Mn(CO)3} moiety is stabilized by an [β-BiW9O33]9− subunit in the “supporting” structural motif, as shown in Fig. 1d. Both Mn centers exhibit octahedral MnO3C3 coordination defined by three oxygen atoms from [β-BiW9O33]9− fragment and three carbon atoms from carbonyl groups (Mn–O: 2.043(1)–2.093(3) Å, Mn–C: 1.785(8)–1.815(7) Å). This supporting model is very similar to those of [(H2M8O30){Mn(CO)3}2]8− polyoxoanions (M = Mo, W)6d but with slight difference (Fig. 1d and e). In [(H2M8O30){Mn(CO)3}2]8−, three μ3-O atoms are edge-sharing oxygen atoms from one M3O9 triad, while the three μ3-O atoms in 1a are one edge-sharing oxygen atoms and two corner-sharing oxygen atoms from two W3O9 triads, which is uncommon in PMCDs. The MnO3C3 moieties, as reflected in M−C distances and M–C–O angle variations, are slightly more distorted from pure octahedral symmetry than the Mn derivatives. Specifically, the Mn–C bond distances are 1.798 Å on average and the Mn–C–O angles are 173.5°, which are similar to those in analogous complexes.
Polyanion 2a exhibits a structure very similar to that of 1a. The main differences are (i) the hetero element in 2a is Sb instead of Bi in 1a and (ii) the adjacent cluster anions in 2a are linked alternately by two symmetry-equivalent [Na3(H2O)12]3+ clusters into a “S-type” one-dimensional (1D) chain (Fig. 2) through Na1–O34–C and Na3–O36–C bridges [Na–O: 2.396(9)–2.696(12) Å]. Apparently, Na1, Na2 and Na3 ions reside in different coordination environments. Na1 and Na3 ions linked each other with one μ2-O7W in the corner-sharing mode, while Na1 and Na2 ions were joined together by three μ2-OW atoms in the face-sharing mode.
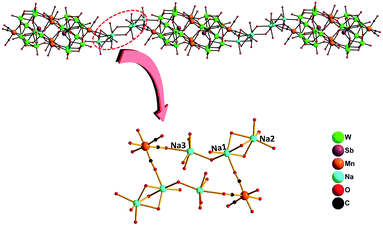 |
| | Fig. 2 Ball-and-stick representation of the quasi-1D chain of 2a along the a-axis. Colour code: O, red balls; C, black balls; Mn, orange balls; W, green balls; Sb, purple balls; Na, turquoise balls. | |
The structure of polyanions 3a and 4a is most closely related to that of 1a. However, each inner manganese center shows crystallographic positional disorder (0.75:0.25) of MnII and WIV (Fig. 3). In addition, the hetero element in 3a is Sb instead of Bi in 1a, while that in 4a is still Bi. Finally, it has to be noted that special structure features of polyanions 3a and 4a, compared with respective situ precursors 5 and 6, are shown in Fig. 3. For polyanion 3a, except the change of attachment the organic moieties onto the POMs skeletons, the transformation of [B-α-SbW9O33]9− → [B-β-SbW9O33]9− by the 60° rotation of one W3O13 in polyanion of 5 occurs as well as the substitution of the tetra-MnII cluster {Mn4(H2O)10}8+ cluster for tri-MnII cluster {Mn(H2O)3}6+ is sealed into the sandwich position of two [B-β-SbW9O33]9−. In comparison with polyanion 6, two distinctions are demonstrated in polyanion 4a. Firstly, on the position of sandwich in 4a framework, two W ions were substituted by two disordered Mn/W ions in the tetra-MnII clusters likely that of 3a. Secondly, two {Mn(CO)3} groups are grafted on both ends up and down of M4Bi2W18 skeleton.
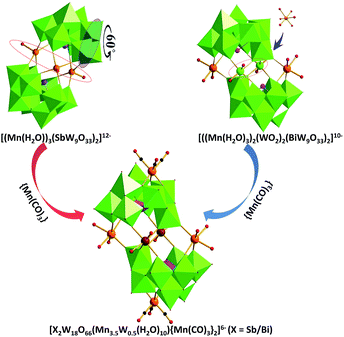 |
| | Fig. 3 Ball-and-stick and polyhedral representations of 3a and 4a, highlighting the structure transition from precursors 5 and 6. Colour code: O, red balls; C, black balls; Mn, orange balls; W, green balls; Mn3.5W0.5, brown balls; Sb/Bi, purple balls; WO6 octahedra, green; SbO3/BiO3 tetrahedra, purple. | |
The bond valence sum calculations indicate that all W, Sb, and Bi atoms in four complexes are in the +6, +3 and +3 oxidation states, respectively. Four Mn atoms at the belt position are in the +2 oxidation states. According to the charge balance consideration, five protons are needed to compensate the negative charges of 1a. However, the bond valence sum calculations of all the oxygen atoms in this compound indicate that all of these protons are delocalized, these protons cannot be located crystallographically, which is common in POM chemistry.11
IR spectra
The IR spectra were recorded in the range of 4000–450 cm−1 for compounds 1–4 and Na9[SbW9O33]·19.5H2O (Fig. S3†). Compared to the IR spectrum of Na9[SbW9O33]·19.5H2O, all these clusters show a fingerprint region characteristic for the tungsten-oxo framework, indicating the presence of Keggin-type trilacunary fragment (Fig. 4). The bands in the range of 948–936 cm−1 are associated with the antisymmetric stretching vibrations of the terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, whereas the bands in the range of 820–814 and 700–687 cm−1 can be mainly attributed to the antisymmetric stretching vibrations of the corner-sharing W–Ob and edge-sharing W–Oc bonds.
O bonds, whereas the bands in the range of 820–814 and 700–687 cm−1 can be mainly attributed to the antisymmetric stretching vibrations of the corner-sharing W–Ob and edge-sharing W–Oc bonds.
 |
| | Fig. 4 IR spectra of 1–4 and Na9[SbW9O33]·19.5H2O in the range of 2250–450 cm−1. | |
Interestingly, the IR spectra of 1–4 are similar to that of the [X2W22O74(OH)2]n−,12 confirming that the compounds 1–4 retain the well-known Krebs' sandwich structure. The largish shifts or splitting in characteristic peaks may be caused by all internal and external tungsten atoms of the polyanion [X2W22O74(OH)2]n− replaced by four Mn2+ ions. In addition, the strong absorption bands at 2030 cm−1 and 1927 cm−1 arise from symmetric and antisymmetric stretching vibration of the C–O bond, respectively. In comparison with the terminal CO position of raw material Mn(CO)5Br, the peaks in these four complexes display obviously red shift, which may attribute to the combination of carbonyl metal groups with POMs frameworks. This is in satisfactory agreement with the solid-state structure.
Electrochemical and electrocatalytic properties
In order to survey the electrochemical behaviors and electrocatalytic properties of compounds 1 and 2, the cyclic voltammetry (CV) experiments were carried out in the mixed solvent Na2SO4/CH3CN (0.4 mol·L−1) (3/1, v/v) at the potential range of −1.5 to +1.2 V. Both compounds display very similar electrochemical behaviors (Fig. 5) and electrocatalytic properties (Fig. S5†), which might result from the similar building units [(Mn2(H2O)5)(XW9O33){Mn(CO)3}]2n−. As shown in Fig. 5, the redox peaks of WVI centers fragments appear at a more negative potential region than that assigned to the MnII centers, as expected. In the negative potential domain, the cyclic voltammogram exhibits one pair of redox waves (III/III′) and two irreversible reduction peaks (I, II), which are attributed to the WVI-centers redox. The ΔEp values of the waves (III/III′) in the cyclic voltammogram are 99 mV for 1 and 153 mV for 2 respectively, which suggest the quasi-reversible redox WVI centers in the POM frameworks.10,13 In the positive potential direction, two pairs of redox waves (IV/IV′ and VI/VI′) and an ill-defined oxidation peak (V) are observed in cyclic voltammograms of 1 and 2, which are assigned to MnII centers redox.14 Thereinto, the wave IV/IV′ and VI/VI′ shows a large difference in peak potential (Table 3), which indicates that the electron transfer is very slow.15 A slow electron transfer step IV/IV′ can be easily assigned to the MnII/MnIII redox step; the second electron transfer VI/VI′ is assigned to the MnIII/MnIV redox step.14 With the scan rates increasing, the corresponding cathodic and anodic peaks slightly increases, but the mean peak potential does not change distinctly. Below 200 mV s−1, the peak currents intensity for the Mn(II) of 1 and 2 redox processes are linearly proportional to the scan rates (Fig. 5), indicating that the redox process are probably surface-controlled in a specific range of scan rates. Furthermore, the electrocatalytic behaviors of both compounds for nitrite were investigated under the same conditions (Fig. S5†). Obviously, with the addition of modest amounts of sodium nitrite, an irreversible oxidation peak appears in the positive range at Epc = 0.85–0.90 V, and this is expected for NO2−.5e Meanwhile, the reduction peak current of the WVI-based wave increase progressively, which indicates that complex 1 and 2 exhibit well electrocatalytic activity toward the reduction of nitrite.16
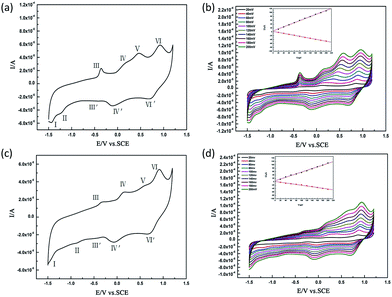 |
| | Fig. 5 (a) CV of 1 at scan rate of 100 mV s−1; (b) CV curves of 1 at different scan rates. Inset figure: the variation of the peak current intensity for the Mn(II) is proportional to the scan rates from 20 to 200 mV s−1 of 1; (c) CV of 2 at scan rate of 100 mV s−1; (d) CV curves of 2 at different scan rates. Inset figure: the variation of the peak current intensity for the Mn(II) is proportional to the scan rates from 20 to 200 mV s−1 of 2. All concentrations above were 1 × 10−4 mol L−1 in CH3CN–Na2SO4 (0.4 mol L−1) (1:3, volume ratio). The working electrode is glassy carbon (3 mm) and the reference electrode is SCE. | |
Table 3 Comparison of voltammetric data for the MnII centers redox of compounds 1 and 2a
| |
Compound 1 |
Compound 2 |
| IV/IV′ |
VI/VI′ |
IV/IV′ |
VI/VI′ |
| [E1/2 = (Epa + Epc)/2, ΔEp = Epa − Epc]. |
| E1/2 (V) |
0.044 |
0.818 |
0.046 |
0.805 |
| ΔEp (V) |
0.264 |
0.207 |
0.198 |
0.217 |
Magnetic properties
The magnetic properties for compounds 1–4 were measured on the polycrystalline samples in 1.8–300 K under a 2k Oe applied field. Their plots of χM and χMT versus T are illustrated in Fig. 6. For the compound 2, the χMT of 17.74 emu K mol−1 at 300 K is in reasonably good agreement with the theoretical value of 17.51 emu K mol−1 calculated for four isolated-high spin Mn2+. Upon cooling, the χMT product decreases tardily to 17.05 emu K mol−1 at 50 K, while below 50 K, the χMT undergoes a sudden drop with temperature and reaches a minimum of 7.56 emu K mol−1 at 1.8 K, which might be assigned to the hydration Mn(II) cluster intermolecular antiferromagnetic interactions. What's more, the magnetic susceptibility follows the Curie–Weiss law over the entire temperature range with C = 18.22 emu K mol−1 and θ = −3.51 K (Fig. 6b). The negative Weiss constant further confirms the presence of the dominating antiferromagnetic interactions between spin carriers.17,18 It is worth mentioning that, for compound 3, the room temperature χMT value is 16.12 emu K mol−1 (Fig. 6c), which is slightly lower than the expected value 17.50 emu K mol−1 calculated for four isolated Mn2+. This characteristic thermal behavior might be the reason of the disorder of manganese atom and tungsten atom. Similarly, the compounds 1 and 4 show the similar thermal behaviors as the compounds 2 and 3. The magnetic data for four compounds in the form of χM, χMT and χM−1 (inset) vs. T plots are presented in Fig. 6.
 |
| | Fig. 6 The plots of χM, χMT versus T for 1 (a), 2 (b), 3 (c) and 4 (d) between 1.8 and 300 K. | |
Experimental section
Materials and instrumentation
All other chemicals were reagent grade and used as purchased without purification. Na9[XW9O33]·19.5H2O (X = Sb/Bi),12,19a Na11(NH4)[(Mn(H2O))3(SbW9O33)2]·45H2O, and Na6(NH4)4[(Mn(H2O)3)2(WO2)2(BiW9O33)2]·37H2O19b were synthesized as previously described. FT-IR spectra were measured on a Bruker VERTEX 70 IR spectrometer using KBr pellets in the range of 4000–500 cm−1. Elemental analyses (C, H and N) were conducted on a Perkin-Elmer 2400-II CHNS/O analyzer. Inductively coupled plasma (ICP) spectra were obtained on a Perkin-Elmer Optima 2000 ICP-OES spectrometer. X-ray powder diffraction (XPRD) spectral data were recorded on a Bruker AXS D8 Advance diffractometer with Cu Kα radiation in the angular range 2θ = 5–45° at 293 K. UV spectra were obtained with a U-4100 spectrometer at room temperature. All electrochemical measurements were performed at room temperature in a standard three-electrode cell connected to a LK98 microcomputer-based electrochemical system. A freshly cleaned glassy carbon disk electrode (3 mm diameter) was used as a working electrode, a platinum wire served as the counter electrode and SEC as the reference electrode. Magnetic susceptibility measurements were carried out with a Quantum Design MPMS-XL-7 magnetometer in the temperature range of 1.8–300 K. The susceptibility data were corrected from the diamagnetic contributions as deduced by using Pascal's constant tables.
Synthesis of compound 1. Mn(CO)5Br (70 mg, 0.25 mmol) in 4 mL CH3CN was refluxed in the dark for 30 min and cooled to room temperature, then dropped into a solution of Na9[BiW9O33]·16H2O (720 mg, 0.25 mmol), Mn(Ac)2·4H2O (123 mg, 0.502 mmol) and NH4NO3 (80 mg, 1 mmol) in 15 mL distilled water at pH value about 6.5. The resulting solution was stirred at 80 °C for 40 min, then 1 mL tetramethylammonium chloride (0.4 M) was added to the solution while hot, then cooled and filtered. The filtrate was allowed to stand in the dark for slow evaporation. Yellow block crystals of 1 were isolated after two weeks. Yield: 48 mg (6.32% based on Bi). Anal. calcd (%) for C6H99Bi2Mn6N3O113W18: C, 1.19; H, 1.64; N, 0.69; Bi, 6.88; W, 54.44; Mn, 5.42; found: C, 2.67; H, 1.88; N, 0.46; Bi, 4.19; W, 55.3; Mn, 5.67. FT-IR (KBr pellet): 3431 (s), 2031 (vs), 1925 (vs), 1631 (m), 1484 (m), 937 (s), 816 (s), 723 (w), 689 (s), 627 (w), 509 (w) cm−1.
Synthesis of compound 2. This compound was prepared using a similar procedure to 1 except that Na9[BiW9O33]·16H2O was replaced by the Na9[SbW9O33]·19.5H2O (716 mg, 0.25 mmol) as the raw material. Yellow block crystals of 2 were isolated after two weeks. Yield 76 mg (9.8% based on Sb). Anal. calcd (%) for C14H116Mn6N6O118Sb2W18: C 2.70; H, 1.88; N, 0.45; Sb, 3.91; W, 53.19; Na, 2.22; Mn, 5.30; found: C, 2.68; H, 2.11; N, 0.46; Sb, 4.19; W, 55.3; Na, 2.14; Mn, 5.67. FT-IR (KBr, pellet): 3433 (vs), 2031 (vs), 1931 (vs), 1631 (m), 1487 (m), 942 (vs), 818 (s), 723 (w), 695 (s), 631 (w), 550 (w) cm−1.
Synthesis of compound 3. Mn(CO)5Br (70 mg, 0.25 mmol) in 3 mL CH3CN was refluxed in the dark for 20 min and cooled to room temperature, then dropped into a solution of Na11(NH4)[(Mn(H2O))3(SbW9O33)2]·45H2O (228 mg, 0.039 mmol) in 15 mL distilled water at pH value about 6.26. The resulting solution was stirred at 80 °C for 40 min, then 1 mL 0.4 M tetramethylammonium chloride was added to the solution while hot, cooled and filtered. The filtrate was allowed to stand in the dark for slow evaporation. Yellow block crystals of 3 were isolated after two weeks. Yield: 56 mg (24.01% based on Sb). Anal. calcd (%) for C14H92Mn7N2O106Sb2W19: C, 2.78; H, 1.53; N, 0.46; Sb, 4.03; W, 56.30; Mn, 6.82; found: C, 2.58; H, 1.52; N, 0.39; Sb, 4.21; W, 56.9; Mn, 6.18. FT-IR (KBr pellet): 3433 (s), 2030 (vs), 1927 (vs), 1630 (m), 1485 (m), 942 (s), 817 (s), 723 (w), 691 (s), 631 (w), 546 (w) cm−1.
Synthesis of compound 4. The procedure is similar to that for preparing 3, but Na11(NH4)[(Mn(H2O))3(SbW9O33)2]·45H2O were replaced by Na6(NH4)4[(MnII(H2O)3)2(WO2)2(BiW9O33)2]·37H2O (0.228 g, 0.049 mmol). Yellow block crystals of 4 were isolated after two weeks. Yield: 47 mg (15.43% based on Bi). Anal. calcd (%). for C14H92Bi2Mn7N2O106W19: C, 2.71; H, 1.49; N, 0.45; Bi, 6.72; W, 54.72; Mn, 6.63; found: C, 2.58; H, 1.52; N, 0.41; Bi, 6.82; W, 55.27; Mn, 6.38. FT-IR (KBr pellet): 3430 (s), 2030 (vs), 1920 (vs), 1630 (m), 1484 (m), 937 (s), 816 (s), 724 (w), 687 (s), 626 (w), 509 (w) cm−1.
X-ray crystallography
Suitable single crystals of 1, 2, 3 and 4, were selected from their respective mother liquors and airproofed in a glass tube. X-ray diffraction intensity data were recorded on a Bruker APEX-II CCD diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). Routine Lorentz and polarization corrections were applied. The absorption correction was based on multiple and symmetry equivalent reflections in the data set using the SADABS program. The structure was solved by direct methods and refined using full-matrix least squares on F2. The crystal was kept at 296(2) K during data collection. Using Olex2,20 the structure was solved with the SHELXS-97 (ref. 21) structure solution program using Direct Methods and refined with the SHELXL-14 (ref. 22) refinement package using Least Squares minimisation. No hydrogen atoms associated with the water molecules were located from the difference Fourier map. A summary of the crystal data and structure refinements is listed in Table 2.
Conclusions
In this work, we have shown that the reaction of [XW9O33]9− (X = Sb/Bi), Mn(CO)5Br and Mn2+ can afford novel organic–inorganic hybrids Mn4-substituted Keggin-type PMCDs, which can be also isolated by the reaction of [{Mn(H2O)}3(SbW9O33)2]12−/[(Mn(H2O)3)2(WO2)2(BiW9O33)2]10− and Mn(CO)5Br. The electrochemical experiments have shown that compounds 1 and 2 exhibit good electrocatalytic activities for the reduction of NO2−. Magnetic measurements on four complexes suggest that the MnII ions are strongly antiferromagnetically coupled. Most importantly, our first attempts to introduce transition metal into PMCD chemistry have shown promising results.
Acknowledgements
We gratefully acknowledge financial support from the Natural Science Foundation of China (21401042), Postdoctoral Science Foundation of Henan Province (2015031), and 2015 Young Backbone Teachers Foundation from Henan Province (2015GGJS-017).
References
-
(a) X.-B. Han, Y.-G. Li, Z.-M. Zhang, H.-Q. Tan, Y. Lu and E.-B. Wang, J. Am. Chem. Soc., 2015, 137, 5486 CrossRef CAS PubMed;
(b) L. Huang, S.-S. Wang, J.-W. Zhao, L. Cheng and G.-Y. Yang, J. Am. Chem. Soc., 2014, 136, 7637 CrossRef CAS PubMed;
(c) Y. Nishimoto, D. Yokogawa, H. Yoshikawa, K. Awaga and S. Irle, J. Am. Chem. Soc., 2014, 136, 9042 CrossRef CAS PubMed;
(d) H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268 CrossRef CAS PubMed;
(e) C. L. Hill, Chem. Rev., 1998, 98, 1 CrossRef CAS PubMed.
-
(a) P. Gouzerh and A. Proust, Chem. Rev., 1998, 98, 101 CrossRef;
(b) A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837 RSC;
(c) W. H. Knoth, J. Am. Chem. Soc., 1979, 101, 2211 CrossRef CAS;
(d) W. H. Knoth, J. Am. Chem. Soc., 1979, 101, 759 CrossRef CAS;
(e) S. Bareyt, S. Piligkos, B. Hasenknopf, P. Gouzerh, E. Lacôt, S. Thorimbert and M. Malacria, J. Am. Chem. Soc., 2005, 125, 6788 CrossRef PubMed;
(f) S. Bareyt, S. Piligkos, B. Hasenknopf, P. Gouzerh, E. Lacôte, S. Thorimbert and M. Malacria, J. Am. Chem. Soc., 2005, 127, 6788 CrossRef CAS PubMed;
(g) U. Kortz, F. Hussain and M. Reicke, Angew. Chem., Int. Ed., 2005, 44, 3773 CrossRef CAS PubMed;
(h) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009 CrossRef CAS PubMed;
(i) S. J. Pastine, D. V. Gribkov and D. Sames, J. Am. Chem. Soc., 2006, 128, 14220 CrossRef CAS PubMed.
- P. Gouzerh and A. Proust, Chem. Rev., 1998, 98, 77 CrossRef CAS PubMed.
-
(a) C. J. Besecker and W. G. Klemperer, J. Am. Chem. Soc., 1980, 25, 7598 CrossRef;
(b) V. W. Day, M. F. Fredrich, M. R. Thompson, W. G. Klemperer, R. S. Liu and W. J. Shum, J. Am. Chem. Soc., 1981, 103, 3597 CrossRef CAS;
(c) C. J. Besecker, V. W. Day, W. G. Klemperer and M. R. Thompson, Inorg. Chem., 1985, 24, 44 CrossRef CAS;
(d) W. G. Klemperer and D. J. Main, Inorg. Chem., 1990, 29, 2355 CrossRef CAS;
(e) W. G. Klemperer and B. Zhong, Inorg. Chem., 1993, 32, 5821 CrossRef CAS;
(f) A. V. Besserguenev, M. H. Dickman and M. T. Pope, Inorg. Chem., 2001, 40, 2582 CrossRef CAS PubMed;
(g) J. Li, J. Guo, J. Jia, P. Ma, D. Zhang, J. Wang and J. Niu, Dalton Trans., 2016, 45, 6726 RSC.
-
(a) W. H. Knoth, J. Am. Chem. Soc., 1979, 101, 2211 CrossRef CAS;
(b) P. J. Domaille and W. H. Knoth, Inorg. Chem., 1983, 22, 818 CrossRef CAS;
(c) A. R. Siedle, W. B. Gleason, R. A. Newmark, R. P. Skarjune, P. A. Lyon, C. G. Markell, K. O. Hodgson and A. L. Roe, Inorg. Chem., 1990, 29, 1667 CrossRef CAS;
(d) M. Sadakane, Y. Iimuro, D. Tsukuma, B. S. Bassil, M. H. Dickman, U. Kortz, Y. Zhang, S. Ye and W. Ueda, Dalton Trans., 2008, 47, 6692 RSC;
(e) J. Zhao, J. Zhao, P. Ma, J. Wang and J. Niu, J. Mol. Struct., 2012, 1019, 61 CrossRef CAS;
(f) J. Zhao, J. Wang, J. Zhao, P. Ma, J. Wang and J. Niu, Dalton Trans., 2012, 41, 5832 RSC;
(g) C. Zhao, C. S. Kambara, Y. Yang, A. L. Kaledin, D. G. Musaev, T. Q. Lian and C. L. Hill, Inorg. Chem., 2013, 52, 671 CrossRef CAS PubMed;
(h) Y. Zhang, D. Zhang, Z. Huo, P. Ma, J. Niu and J. Wang, RSC Adv., 2014, 4, 28848 RSC;
(i) Z. Huo, J. Zhao, Z. Bu, P. Ma, Q. Liu, J. Niu and J. Wang, ChemCatChem, 2014, 6, 3096 CrossRef CAS;
(j) C. Zhao, E. N. Glass, J. M. Sumliner, J. Bacsa, D. T. Kim, W. Guo and C. L. Hill, Dalton Trans., 2014, 43, 4040 RSC;
(k) Z. Huo, J. Guo, J. Lu, Q. Xu, P. Ma, J. Zhao, D. Zhang, J. Niu and J. Wang, RSC Adv., 2015, 5, 69006 RSC;
(l) Y. Liu, Y. Zhang, P. Ma, Y. Dong, J. Niu and J. Wang, Inorg. Chem. Commun., 2015, 56, 45 CrossRef CAS.
-
(a) J. Wang, J. Li and J. Niu, Chem. Res. Chin. Univ., 2008, 24, 675 CAS;
(b) R. Villanneau, R. Delmont, A. Proust and P. Gouzerh, Chem.–Eur. J., 2000, 6, 1184 CAS;
(c) J. Niu, L. Yang, J. Zhao, P. Ma and J. Wang, Dalton Trans., 2011, 40, 8298 RSC;
(d) D. Zhang, J. Zhao, Y. Zhang, P. Ma, J. Wang and J. Niu, Dalton Trans., 2013, 42, 2696 RSC.
-
(a) T. Nagata, M. Pohl, H. Weiner and R. G. Finke, Inorg. Chem., 1997, 36, 1366 CrossRef CAS PubMed;
(b) C. Zhao, Z. Huang, W. Rodríguez-Córdoba, C. S. Kambara, K. P. O'Halloran, K. I. Hardcastle, D. G. Musaev, T. Lian and C. L. Hill, J. Am. Chem. Soc., 2011, 133, 20134 CrossRef CAS PubMed;
(c) C. Zhao, W. Rodríguez-Córdoba, A. L. Kaledin, Y. Yang, Y. V. Geletii, T. Lian, D. G. Musaev and C. L. Hill, Inorg. Chem., 2013, 52, 13490 CrossRef CAS PubMed.
-
(a) J. Liu, Q. Han, L. Chen and J. Zhao, CrystEngComm, 2016, 18, 842 RSC;
(b) S.-T. Zheng and G.-Y. Yang, Chem. Soc. Rev., 2012, 41, 7623 RSC;
(c) W. Guo, H. Lv, J. Bacsa, Y. Gao, J.
S. Lee and C. L. Hill, Inorg. Chem., 2016, 55, 461 CrossRef CAS PubMed.
-
(a) W.-L. Chen, B.-W. Chen, H.-Q. Tan, Y.-G. Li, Y.-H. Wang and E.-B. Wang, J. Solid State Chem., 2010, 183, 310 CrossRef CAS;
(b) L.-H. Bi, U. Kortz, S. Nellutla, A. C. Stowe, J. Tol, N. S. Dalal, B. Keita and L. Nadjo, Inorg. Chem., 2005, 44, 896 CrossRef CAS PubMed;
(c) N. Laronze, J. Marrot and G. Herve, Inorg. Chem., 2003, 42, 5857 CrossRef CAS PubMed;
(d) Z. Zhang, Q. Lin, S.-T. Zheng, X. Bu and P. Feng, Chem. Commun., 2011, 47, 3918 RSC.
-
(a) U. Kortz, M. G. Savelieff, B. S. Bassil, B. Keita and L. Nadjo, Inorg. Chem., 2002, 41, 783 CrossRef CAS PubMed;
(b) M. Carraro, B. S. Bassil, A. Sorarù, S. Berardi, A. Suchopar, U. Kortz and M. Bonchio, Chem. Commun., 2013, 49, 7914 RSC.
-
(a) P. Mialane, A. Dolbecq, L. Lisnard, A. Mallard, J. Marrot and F. Sécheresse, Angew. Chem., Int. Ed., 2002, 41, 2398 CrossRef CAS;
(b) H.-Y. An, E.-B. Wang, D.-R. Xiao, Y.-G. Li, Z.-M. Su and L. Xu, Angew. Chem., Int. Ed., 2006, 45, 904 CrossRef CAS PubMed;
(c) E. M. Villa, C. A. Ohlin, E. Balogh, T. M. Anderson, M. D. Nyman and W. H. Casey, Angew. Chem., Int. Ed., 2008, 47, 4844 CrossRef CAS PubMed.
-
(a) M. Bosing, I. Loose, H. Pohlmann and B. Krebs, Chem.–Eur. J., 1997, 3, 1232 CrossRef CAS;
(b) I. Loose, E. Droste, M. Bosing, H. Pohlmann, M. H. Dickman, C. Rosu, M. T. Pope and B. Krebs, Inorg. Chem., 1999, 38, 2688 CrossRef CAS.
-
(a) L.-H. Bi, T. McCormac, S. Beloshapkin and E. Dempsey, Electroanalysis, 2008, 20, 38 CrossRef CAS;
(b) J. Thiel, C. Ritchie, H. N. Miras, C. Streb, S. G. Mitchell, T. Boyd, M. N. C. Ochoa, M. H. Rosnes, J. McIver, D.-L. Long and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 6984 CrossRef CAS PubMed;
(c) B. S. Bassil, M. Ibrahim, S. S. Mal, A. Suchopar, R. N. Biboum, B. Keita, L. Nadjo, S. Nellutla, J. V. Tol, N. S. Dalal and U. Kortz, Inorg. Chem., 2010, 49, 4949 CrossRef CAS PubMed;
(d) U. Kortz, M. G. Savelieff, B. S. Bassil, B. Keita and L. Nadjo, Inorg. Chem., 2002, 41, 783 CrossRef CAS PubMed;
(e) Y. Huo, D. Li, R. Wan, P. Ma, D. Zhang, J. Niu and J. Wang, RSC Adv., 2015, 5, 106077 RSC;
(f) J.-W. Zhao, J. Cao, Y.-Z. Li, J. Zhang and L.-J. Chen, CrystEngComm, 2014, 14, 6217 CAS.
-
(a) M. Lebrini, I. M. Mbomekallé, A. Dolbecq, J. Marrot, P. Berthet, J. Ntienoue, F. Sécheresse, J. Vigneron and A. Etcheberry, Inorg. Chem., 2011, 50, 6437 CrossRef CAS PubMed;
(b) B. Keita, P. Mialane, F. Sécheresse, P. de Oliveira and L. Nadjo, Electrochem. Commun., 2007, 9, 164 CrossRef CAS.
- B.-H. Zhu, L. Zhang, N. Xiao, J.-B. Chen, Y.-Q. Yi and J. Sun, Inorg. Chim. Acta, 2004, 357, 864 CrossRef CAS.
-
(a) Y.-W. Li, Y.-G. Li, Y.-H. Wang, X.-J. Feng, Y. Lu and E.-B. Wang, Inorg. Chem., 2009, 48, 6452 CrossRef CAS PubMed;
(b) L.-H. Bi, G. Al-Kadamany, E. V. Chubarova, M. H. Dickman, L. F. Chen, D. S. Gopala, R. M. Richards, B. Keita, L. Nadjo, H. Jaensch, G. Mathys and U. Kortz, Inorg. Chem., 2009, 48, 10068 CrossRef CAS PubMed;
(c) L. Chen, J. Cao, X. Li, X. Ma, J. Luo and J. Zhao, CrystEngComm, 2015, 17, 5002 RSC;
(d) C. Pichon, P. Mialane, A. Dolbecq, J. Marrot, E. Rivière, B. Keita, L. Nadjo and F. Sécheresse, Inorg. Chem., 2007, 46, 5292 CrossRef CAS PubMed.
-
(a) Q. Wu, Y.-G. Li, Y.-H. Wang, E.-B. Wang, Z.-M. Zhang and R. Clérac, Inorg. Chem., 2009, 48, 1606 CrossRef CAS PubMed;
(b) H.-H. Wu, S. Yao, Z.-M. Zhang, Y.-G. Li, Y. Song, Z.-J. Liu, X.-B. Han and E.-B. Wang, Dalton Trans., 2013, 42, 342 RSC;
(c) R. Sato, K. Suzuki, T. Minato, M. Shinoe, K. Yamaguchi and N. Mizuno, Chem. Commun., 2015, 51, 4081 RSC.
-
(a) J. Vallejo, A. Pascual-Álvarez, J. Cano, I. Castro, M. Julve, F. Lloret, J. Krzystek, G. De Munno, D. Armentano, W. Wernsdorfer, R. Ruiz-García and E. Pardo, Angew. Chem., Int. Ed., 2013, 52, 14075 CrossRef CAS PubMed;
(b) R. Ishikawa, R. Miyamoto, H. Nojiri, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2013, 52, 8300 CrossRef CAS PubMed;
(c) A. Grigoropoulos, M. Pissas, P. Papatolis, V. Psycharis, P. Kyritsis and Y. Sanakis, Inorg. Chem., 2013, 52, 12869 CrossRef CAS PubMed;
(d) G. A. Craig, J. J. Marbey, S. Hill, O. Roubeau, S. Parsons and M. Murrie, Inorg. Chem., 2015, 54, 13 CrossRef CAS PubMed;
(e) L. Chen, J. Wang, Y.-Z. Liu, Y. Song, X.-T. Chen, Y.-Q. Zang and Z.-L. Xue, Eur. J. Inorg. Chem., 2015, 2, 271 CrossRef.
-
(a) B. Botar, T. Yamase and E. Ishikawa, Inorg. Chem. Commun., 2000, 3, 579 CrossRef CAS;
(b) M. Bosing, A. Noh, I. Loose and B. Krebs, J. Am. Chem. Soc., 1998, 120, 7252 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The summary of Keggin-type PMCDs, IR spectra, the XRPD patterns, UV-vis spectrum and the CV of 1–4. CCDC 1505560–1505563. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra23547f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.