
Synthesis and application of magnetic reduced graphene oxide composites for the removal of bisphenol A in aqueous solution—a mechanistic study†
 *abc
*abc
Abstract
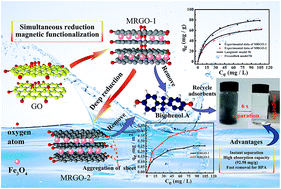
Two kinds of magnetic reduced graphene oxide composites (MRGO) namely MRGO-1 and MRGO-2 with different reduction degrees are synthesized by a facile method. MRGO-2 is obtained by further reduction of MRGO-1 using hydrazine to obtain a relatively deeper reduction degree. Removal of bisphenol A (BPA) from aqueous solution using MRGO-1 and MRGO-2 is investigated. The kinetics and isotherm data of BPA absorbed by MRGO-1 and MRGO-2 are both well fitted with the pseudo-second-order kinetic model and the Langmuir isotherm, respectively. The maximum adsorption capacity of MRGO-1 and MRGO-2 for BPA obtained from the Langmuir isotherm is 92.98 mg g−1 and 71.66 mg g−1 at 288.15 K, respectively. Furthermore, the used MRGO-1 and MRGO-2 could be collected and recycled efficiently via an external magnet. The BPA adsorption capacity of the MRGO-1 still remained greater than 86% of the initial adsorption capacity after nine repeated absorption–desorption cycles. A thermodynamic study shows that the adsorption is a spontaneous and exothermic process. The maximum adsorption capacity of MRGO-1 for BPA is higher than that of MRGO-2, implying a deeper reduction process to MRGO-2 from MRGO-1 is not essential for the removal of BPA from aqueous solution. The energy is saved by omitting the deep reduction process. It is also found that if severe aggregation of graphene sheets in the deep reduction process to MRGO-2 from MRGO-1 can be alleviated, the corresponding absorption capacity of MRGO-2 for BPA may be greatly improved.
 Please wait while we load your content...
Please wait while we load your content...

