
Understanding the significance of O-doped graphene towards biomedical applications†
Shrikant S. Maktedar,
Gopal Avashthi and
Man Singh*
School of Chemical Sciences, Central University of Gujarat, Gandhinagar-382030, India. E-mail: mansingh50@hotmail.com; shrikantmaktedar@gmail.com; gopalavashthi@gmail.com; Fax: +91-079-23260076; Tel: +91-079-23260210
First published on 23rd November 2016
Abstract
The inherent stability and solvent processability restrict the functional expression of graphene for biomedical applications. To resolve this problem graphene oxide (GrO) has emerged as a biomaterial for biomedical applications of graphene. The participation of O_K edges has been confirmed with synchrotron radiation near edge X-ray absorption fine structure (SR-NEXAFS). The as-prepared graphene oxide has been confirmed with 13C cross polymerization magic angle spinning (CP-MAS) solid state NMR, HRXPS, HRTEM, SAED, SEM, AFM, Raman, XRD, FTIR, UV, TGA, DTA, BET and Porosimeter, etc. The biomedical significance of graphene oxide has been supported by an extensive structural investigation. The underlying mechanism of biological activity of graphene oxide has been inferred through the synergistic impact of contributory factors (SICF). The cytotoxicity of graphene oxide has been evaluated with an SRB assay with living mammalian cell lines, human breast cancer cell line MCF-7 and monkey normal kidney cell line Vero. The estimated cell viabilities are greater than 80% over a 10–80 μg mL−1 concentration. The high cytocompatibility of graphene oxide has confirmed its low toxicity and hence its excellent biocompatibility. Moreover, the morphological effect on the Vero cell line has been endorsed by in vitro behaviour. The sensitivity of graphene oxide has been evaluated with minimum inhibitory concentration (MIC) against antifungal strain C. albicans (ATCC 10231) at 128 μg mL−1. However, for antibacterial strains S. aureus (ATCC 25923) and E. coli (ATCC 25922) the MIC of graphene oxide is 256 μg mL−1. The genetic variability greatly increases during the sub-culturing of strains; therefore the resistant behaviour of graphene oxide has been observed against NCIM strains. Additionally, the antioxidant activity of graphene oxide is 50.57–45.89%, at over 40–100 μg mL−1 concentration. Hence, the structural investigations of graphene oxide have complemented the findings for biological activity and emerged as an advanced functional biomaterial for biomedical applications of graphene.
1. Introduction
The carbon allotropes fullerene, CNT and graphene have an extraordinary application in all the fields of science due to their numerous dimensionality.1–8 The lower dimensionality of carbon allotropes was a cause of its high surface area and unique strength. Among the carbon allotropes, graphene has a unique 2-D surface with a flat hexagonal structure. The structural significance of graphene oxide has already fortified its potential for biomedical applications.36,60,61 The graphene exists in various forms of carbon in numerous dimensions like 3D in graphite, 2D in graphene, 1D in nanotubes and 0D in fullerene. However, the hydrophobicity of pristine graphene was the biggest obstacle for the exploitation of graphene in biomedical applications. Usually, graphite was used as the precursor for chemical synthesis of graphene. Therefore, the 2D graphene in 3D graphite was also difficult to process further due to its highest stability and insolubility in organic solvents. Hence, graphite (Gt) was first transformed into graphite oxide (GtO) and then into graphene oxide to serve the purpose of surface modification for various applications.9–13 The type of dimensionality is the basic difference between graphene (GE), graphene oxide (GrO) and graphite nanoplatelets (GNPs).14–22 Junwei Gu et al. have reported an improved thermal conductivity of ultra-high molecular weight polyethylene (UHMWPE) with a GNPs–GNPs network.14 Junwei Gu et al. have demonstrated the formation of a highly thermally conducting nanocomposite, GNPs/E-51, with a casting method.15 Recently, Juwei Gu et al. have reported the self-healing performance of a (BADCy/PGMA)/E-51 composite comprising 8 wt% BADCy/PGMA microcapsules.16 Juwei Gu et al. have demonstrated the formation of a POSS-g-nBN/BMI/DABA nanocomposite with 15.4 vol% of POSS-g-nBN and found that it is an ideal dielectric composite material with high thermal conductivity and outstanding thermal stability.17 Hongbo Gu et al. have critically reviewed state-of-the-art multifunctional epoxy nanocomposites with magnetic, electrically conductive, thermally conductive, and flame retardant properties.18 Weifeng Zhao et al. have systematically reported the self-alignment of thermally reduced graphene sheets (TRG) that enable highly efficient heat transfer paths in their poly(p-phenylene benzobisoxazole) (PBO)-based nanocomposite films along the in-plane direction, which was achieved for the first time without any assistance from external magnetic or electric fields.19 Li Zhang et al. have studied electrospun one-dimensional TiO2/graphene oxide (GO) composite nanofiber photocatalysts using polyvinylpyrrolidone (PVP) as a fiberizing carrier.20 Bo Jiang et al. have systematically studied the improved thermal stability of methylsilicone resins by compositing with N-doped graphene oxide/Co3O4 nanoparticles.21 Qingbo Zhang et al. have reported that GO modified sizing agents can improve the interfacial properties of carbon fibers/epoxy composites effectively, with a 1 wt% optimum amount of GO.22Graphene oxide was hydrophilic mainly due to the presence of surplus oxygen-containing functionalities. The hydroxyl, epoxide, kenotic and carboxyl functionalities over the surface of graphene oxide render active reaction sites for functionalization. Still, the structure of graphene oxide is ambiguous.23–31 Extensive research on the structure of graphene oxide infers the position of carboxyl (HO–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at the edges, and hydroxyl (–OH) and epoxide (C–O–C) located on the basal planes close to each other.32,33 Thus, the stoichiometric ratio of the functional groups of graphene oxide varies, but after achieving the threshold oxidation level the structure and composition of graphene oxide remain the same.34 Additionally, the higher surface area of graphene oxide is a boon for biomedical applications. The large surface area acts as a scaffold for targeted drug delivery and tissue engineering applications. Hence, graphene oxide emerged as a better alternative for biomedical applications than graphene.35–49 The hydrophilicity of graphene oxide serves the purpose of functionalization for probing biomedical applications.50–59 However, as compared to graphene oxide, functionalized graphene oxide was less significant for biomedical applications, due to its reduced oxygen functionalities and attachment of organic molecules.60,61 Therefore, the present study exemplifies the significance of oxygen over the surface of graphene through the synergistic impact of contributory factors (SICF). The contribution of high surface area, edge structure, pore and surface morphology, composition, impact of atomic domains, defects etc. was significant for biomedical applications of graphene. Hence, graphene oxide has truly emerged as a functional biomaterial for biomedical applications of graphene.
O) at the edges, and hydroxyl (–OH) and epoxide (C–O–C) located on the basal planes close to each other.32,33 Thus, the stoichiometric ratio of the functional groups of graphene oxide varies, but after achieving the threshold oxidation level the structure and composition of graphene oxide remain the same.34 Additionally, the higher surface area of graphene oxide is a boon for biomedical applications. The large surface area acts as a scaffold for targeted drug delivery and tissue engineering applications. Hence, graphene oxide emerged as a better alternative for biomedical applications than graphene.35–49 The hydrophilicity of graphene oxide serves the purpose of functionalization for probing biomedical applications.50–59 However, as compared to graphene oxide, functionalized graphene oxide was less significant for biomedical applications, due to its reduced oxygen functionalities and attachment of organic molecules.60,61 Therefore, the present study exemplifies the significance of oxygen over the surface of graphene through the synergistic impact of contributory factors (SICF). The contribution of high surface area, edge structure, pore and surface morphology, composition, impact of atomic domains, defects etc. was significant for biomedical applications of graphene. Hence, graphene oxide has truly emerged as a functional biomaterial for biomedical applications of graphene.
2. Experimental
2.1 Materials
Graphite flakes, concentrated sulphuric acid (95–98%), phosphoric acid (>85 wt% in H2O), and concentrated hydrogen chloride, were procured from Sigma-Aldrich. 30% hydrogen peroxide was provided by S.D Fine chemicals. Analytical grade potassium permanganate was supplied by Rankem. Absolute alcohol was obtained from Scvuksmandli Ltd. India. All chemicals were used without further purification.2.2 Synthesis of graphene oxide
Herein, the most abundant, easily available and cheapest 3-D graphite flakes were used as raw material for the wet chemical synthesis of 2-D graphene oxide. Efficient graphite oxide (GtO) with a high degree of oxidation was achieved with slight modifications.10 Briefly, the graphite flakes were oxidized with a mixture of concentrated H2SO4/H3PO4 in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (180:20 mL). The concentrated mixture of H2SO4/H3PO4 was poured into a 500 mL RB flask containing a mixture of graphite flakes (1.5 g, 1 wt equiv.) and KMnO4 (9.0 g, 6 wt equiv.). This addition will produce a slight exothermic reaction of 35–40 °C. The produced exotherm cooled to RT. Then, the oil bath was heated over a magnetic stirrer with a hot plate to maintain a temperature of 50 °C. The RB flask holding the reaction mixture was kept inside the oil bath and heated to 50 °C with constant stirring for 12 h. After 12 h, the reaction was cooled at RT. For the workup, the reaction mixture was poured onto ice-cold water (∼200 mL) with 30% H2O2 (1.5 mL). The ice-cold water with the poured reaction mixture was stirred continuously for ten min until oxidation ceased completely. The reaction mixture was centrifuged at 6000 rpm for 0.5 h, to separate larger unreacted aggregates and the supernatant containing the inorganic salts was decanted away from the sediment. The exhaustive washing of the remaining solid was done thrice. For each washing a succession of 200 mL of water, 150 mL of 30% HCl, and 100 mL of ethanol was used. For each wash the filtrate was centrifuged at 6000 rpm for 0.5 h and the supernatant decanted away. After the multiple-wash process, the remaining material was coagulated with 100 mL of ether. The obtained brownish coloured solid cake was vacuum-dried overnight at 50 °C. The formation of brown coloured GtO is generally reported for well-oxidized GtO.11,12
:1 ratio (180:20 mL). The concentrated mixture of H2SO4/H3PO4 was poured into a 500 mL RB flask containing a mixture of graphite flakes (1.5 g, 1 wt equiv.) and KMnO4 (9.0 g, 6 wt equiv.). This addition will produce a slight exothermic reaction of 35–40 °C. The produced exotherm cooled to RT. Then, the oil bath was heated over a magnetic stirrer with a hot plate to maintain a temperature of 50 °C. The RB flask holding the reaction mixture was kept inside the oil bath and heated to 50 °C with constant stirring for 12 h. After 12 h, the reaction was cooled at RT. For the workup, the reaction mixture was poured onto ice-cold water (∼200 mL) with 30% H2O2 (1.5 mL). The ice-cold water with the poured reaction mixture was stirred continuously for ten min until oxidation ceased completely. The reaction mixture was centrifuged at 6000 rpm for 0.5 h, to separate larger unreacted aggregates and the supernatant containing the inorganic salts was decanted away from the sediment. The exhaustive washing of the remaining solid was done thrice. For each washing a succession of 200 mL of water, 150 mL of 30% HCl, and 100 mL of ethanol was used. For each wash the filtrate was centrifuged at 6000 rpm for 0.5 h and the supernatant decanted away. After the multiple-wash process, the remaining material was coagulated with 100 mL of ether. The obtained brownish coloured solid cake was vacuum-dried overnight at 50 °C. The formation of brown coloured GtO is generally reported for well-oxidized GtO.11,12
The oxygen is very energetic and intercalates into the layers of Gt for GtO formation.36 The XRD spectrum of Gt exactly matches the hexagonal Gt, PDF 00-025-0284. This suggests that the hexagonal Gt layers are arranged in an ABAB sequence, meaning that every second graphene layer is identical. Therefore, the position of all the atoms is the same in the plane in ABAB stacking. The shift in diffraction peak and enhancement in interlayer spacing suggest a higher degree of graphite oxidation. The as-prepared GtO was sonicated with ultrasound for 180 min to obtain the highly exfoliated homogeneous dispersion of graphene oxide. The XRD spectrum confirmed the formation of graphene oxide from Gt (Fig. 1). The (002) plane diffraction peaks in the XRD spectrum of Gt and graphene oxide are observed at 26.28 and 10.54 degrees, respectively. The interlayer spacing in Gt is 3.38 Å. After oxidation the interlayer spacing is enhanced to 8.38 Å owing to intercalation of oxide species between the layers of Gt. The specific surface area of graphene oxide is determined by the BET method (Brunauer–Emmett–Teller) to be 104.41 m2 g−1 (ESI Fig. S1†). Junya Wang et al. have reported the significance of the high surface area of graphene oxide as a high temperature CO2 adsorbent. According to them, nanocomposites with different loadings of graphene oxide have a larger adsorption capacity, which is more than twice as large as that of pure layered double hydroxide (LDH).62 However, the cylindrical morphology of graphene oxide has been determined with a Porosimeter (ESI Fig. S1†). The high surface area and cylindrical morphology of graphene oxide provide strength to its structure. Therefore graphene oxide has acted as a functional biomaterial for various biomedical applications.
 | ||
| Fig. 1 XRD spectrum of (a) graphite (Gt) (b) graphene oxide (GrO). | ||
2.3 Characterization techniques
The synthesis of the above mentioned compounds was carried out with a REMI 1 MLH magnetic stirrer with a hot plate. The purification was carried with an Eltek centrifuge, MP 400. The various functionalities over the synthesized compounds were identified with a PerkinElmer Spectrometer 65 FTIR, and a Bruker 400 MHz NMR spectrometer with a solid probe. The evolved electronic states were characterized with near edge X-ray absorption fine structure (NEXAFS) spectroscopy on the SXAS beamline BL-01 at Indus-2 (Synchrotron radiation source 2.5 GeV, 100 mA), Raja Ramanna Centre for Advanced Technology (RRCAT), Indore. High resolution UHV X-ray photoelectron spectroscopy, Omicron ESCA+ was used for the compositional confirmation. The Al Kα radiation of 1486.7 eV energy was operated at 15 kV and 20 mA, throughout the measurements. The high resolution XPS spectra were collected at pass energy 20. The Raman spectra of the samples were recorded with a Horiba HR 800 instrument with a 480 nm Ar laser. The thermogravimetric stabilities to heat were carried out under ambient N2, measured with an EXSTAR TG/DTA 7300 analyser. The powder XRD spectra were obtained with Cu Kα radiation λ = 1.54 Å operated at 40 kV voltage and 30 mA cathodic current. The spectra were recorded in a range of 5 to 80° at 0.01 degree per s scan rate. The TEM data was collected by using JEOL-2100 with an LaB6 electron gun operated at 200 kV. The SEM images were collected with Evo-18, Carl Zeiss. The AFM measurements were conducted in non-contact mode with XE-76 Park Systems equipment. The surface area was calculated by Brunauer, Emmett and Teller (BET) theory, Thermo Scientific (Surfer). The UV spectrum was recorded with a Spectro 2060 instrument.2.4 SRB assay
The in vitro cytotoxicity evaluation was conducted with SRB assay on mammalian cell lines, human breast cancer cell line MCF-7 and monkey normal kidney cell line Vero.63,64 The cell lines were grown in RPMI 1640 medium containing 10% fetal bovine serum and 2 mM of L-glutamine. For the present screening experiment, cells were inoculated into 96 well microtiter plates in 100 μL at plating densities as shown in the study details, depending on the doubling time of individual cell lines. After cell inoculation, the microtiter plates were incubated at 37 °C, 5% CO2, 95% air and 100% relative humidity for 24 h prior to addition of graphene oxide. Graphene oxide was initially solubilized in dimethyl sulfoxide at 100 mg mL−1 and diluted to 1 mg mL−1 using water and stored frozen prior to use. At the time of graphene oxide addition, an aliquot of frozen concentrate (1 mg mL−1) was thawed and diluted to 100 μg mL−1, 200 μg mL−1, 400 μg L−1 and 800 μg mL−1 with complete medium containing the test article. Aliquots of 10 μL of these different graphene oxide dilutions were added to the appropriate microtiter wells already containing 90 μL of medium, resulting in the required final graphene oxide concentrations i.e. 10 μg mL−1, 20 μg mL−1, 40 μg mL−1, 80 μg mL−1. After graphene oxide addition, the plates were incubated under standard conditions for 48 hours and the assay was terminated by the addition of cold TCA. Cells were fixed in situ by the gentle addition of 50 μL of cold 30% (w/v) TCA (final concentration, 10% TCA) and incubated for 60 minutes at 4 °C. The supernatant was discarded; the plates were washed five times with tap water and air dried. Sulforhodamine B (SRB) solution (50 μL) at 0.4% (w/v) in 1% acetic acid was added to each of the wells, and the plates were incubated for 20 minutes at room temperature. After staining, unbound dye was recovered and the residual dye was removed by washing five times with 1% acetic acid. The plates were air dried. The bound stain was subsequently eluted with 10 mM trizma base, and the absorbance was read on a plate reader at a wavelength of 540 nm with 690 nm reference wavelength. Percentage growth was calculated on a plate-by-plate basis for test wells relative to control wells. Percentage growth was expressed as the ratio of average absorbance of the test well to the average absorbance of the control wells × 100. Using the six absorbance measurements [time zero (Tz), control growth (C), and test growth in the presence of graphene oxide at the four concentration levels (Ti)], the percentage growth was calculated at each of the graphene oxide concentration levels. Percentage growth inhibition was calculated as| [Ti/C] × 100% |
2.5 Antimicrobial assay
Through the tube dilution method, the bactericidal activities of graphene oxide were investigated against human pathogenic Gram-positive (S. aureus; ATCC 25923) and Gram-negative (E. coli; ATCC 25922). However, fungicidal activity was perceived against C. albicans (ATCC 10231). Each step of the microbiological experiment was carried out by using aseptic conditions.65 The bacteria were cultured on Mueller Hinton broth at 37 °C, 7.4 pH for 24 h. Seven sterile capped test tubes were taken for experiment. 3.0 mL of sterile Muller Hilton broth was added to the first tube. To all other tubes 2.0 mL of sterile Muller Hilton broth was added. Ampicillin, gentamicin, and fluconazole were used as the standard drugs for Gram-negative, Gram-positive and fungal strains, respectively. 1.0 mL of (conc. 4.096 mg mL−1) chemical compound was added to the first tube. The final concentration prepared in the first tube was 1.024 mg mL−1. A separate pipette was used to mix the contents of this tube and to transfer 2.0 mL to the 2nd tube. Dilutions continued in this manner to tube number 6, being certain to change pipettes between tubes to prevent carryover of chemical compound on the external surface of the pipette. 2.0 mL was removed from tube 6 and discarded. The seventh tube, which served as a control, received no chemical compound. Several colonies of selected bacteria were suspended in normal saline and a McFarlands 0.5 turbidity suspension was prepared. McFarlands 0.5 turbidity suspension was added to each of the tubes. All tubes were incubated overnight at 37 °C. The lowest concentration (highest dilution) without graphene oxide is the minimal inhibitory concentration (MIC).3. Results and discussion
3.1 Structural investigation
The symmetric structure of graphene oxide was confirmed with Raman spectroscopy.36 The characteristic D and G bands in the Raman spectra of graphene oxide were observed at 1359 and 1596 cm−1, respectively (Fig. 2a). The D peak corresponds to structural defects and A1g symmetry while the G peak corresponds to the graphitic domain of E2g symmetry. The D/G ratio was equal to 0.85, confirming the good degree of oxidation with a large amount of defects within the crystal lattice. The 2D band becomes broader with lower intensity and the hump was observed in the range of 2500–3400 cm−1 (Fig. 2b). This 2D band was the characteristic band of multilayer graphenic domains and confirmed the formation of graphene oxide. | ||
| Fig. 2 Raman spectrum of GrO (a) D and G band (b) 2D band. | ||
The oxidation of graphite and the presence of different functionalities was verified with FTIR spectroscopy.60 The FTIR spectrum of graphene oxide (Fig. 3a) shows the formation of multifunctional graphene oxide, after a good degree of graphite oxidation. The characteristic peaks for these functionalities were observed at ∼3403 cm−1 (–OH), ∼1630 (CC), ∼1072 cm−1 (C–O–C) and ∼1734 cm−1 (CO). The UV-vis spectrum of graphene oxide shows two types of electronic transitions (Fig. 3b). The peak at 230 nm was ascribed to π–π* and the peak at 300 nm corresponds to the n–π* transition.
 | ||
| Fig. 3 (a) FTIR and (b) UV spectrum of GrO. | ||
The structure of graphene oxide was identified with 13C solid state NMR (Fig. 4).60 The 13C SSNMR spectrum of graphene oxide (Fig. 4) exhibits peaks at ∂ = 61 ppm (epoxide), ∂ = 71.87 ppm (alcoholic carbon), ∂ = 133.06 ppm (graphitic sp2 carbon), ∂ = 164 ppm (carbonyl of ester) and ∂ = 190 ppm (ketone carbonyl).
 | ||
| Fig. 4 Solid state NMR spectrum of graphene oxide (GrO). | ||
The composition of graphene oxide was confirmed with XPS (Fig. 5).36 The survey spectra of graphite and graphene oxide have been measured in the range of 0–1000 eV. The survey spectrum of graphene oxide is compared with that of graphite (Fig. 5a). The survey spectrum of graphite is mainly comprised of carbon. In addition to carbon, the survey spectrum of graphene oxide is comprised of oxygen, which confirm the higher degree of graphite oxidation. The double peaks in the high resolution C 1s spectrum of graphene oxide confirm the formation of extremely oxidized graphene oxide (Fig. 5b). The high-resolution C 1s spectrum of graphene oxide was measured with pass energy of 20. The deconvoluted C 1s spectrum of graphene oxide produces four peaks at different binding energies. These four different carbons were observed at 283.2 (CC), 284.3 (CO), 286.4 (CO), and 288.1 eV (O–CO). The % concentrations of these carbons are 11.08, 30.11, 50.03 and 8.78, respectively. The decreasing order of binding energies for graphene oxide carbons is
| O–CO > CO > C–O > CC |
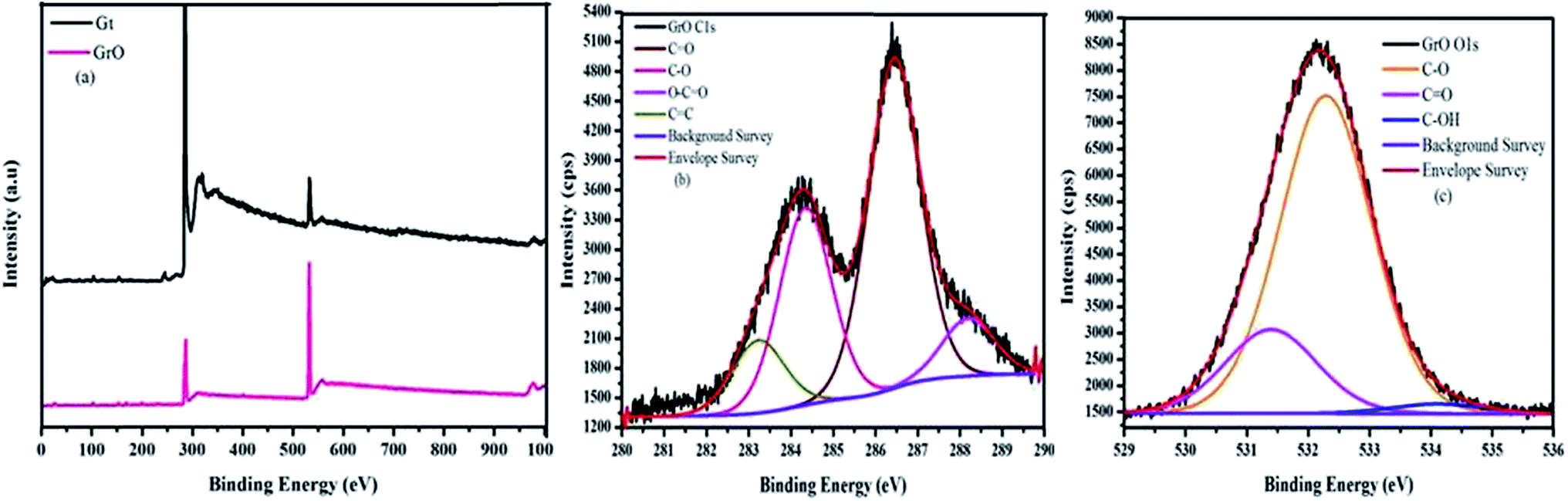 | ||
| Fig. 5 XPS spectrum (a) comparative survey spectrum (b) C 1s spectrum of GrO (c) O 1s spectrum of GrO. | ||
The high resolution O 1s spectra of oxygen confirmed the presence of surplus oxygen-containing functionalities (Fig. 5c). The three deconvoluted peaks in the O 1s spectra of oxygen confirm the presence of three different types of oxygen over the surface of graphene. Therefore, an independent study of 13C solid state NMR and XPS spectra of graphene oxide evidently confirm a good degree of graphite oxidation and formation of graphene oxide.
X-ray absorption spectroscopy (XAS) is a very powerful technique for the determination of electronic transitions. The electronic states of graphene oxide were studied with near edge X-ray absorption fine structure (NEXAFS) at carbon and oxygen K-edges.61 The C_K edge of graphene oxide was compared with reference graphite foil. The comparison of the C_K edge of graphene oxide and graphite is shown in Fig. 6a. The spectra were normalized from 0 to 1 for easy comparison. The newly evolved features between pre and post edge and increased intensity at the C_K edge of graphene oxide evidently confirm the good degree of oxidation. Fig. 6a clearly shows the presence of unoccupied σ* and π* states in graphene oxide. This indicates the restoration of graphitic carbon along with the other functionalities. The σ* and π* states of graphene oxide were observed at 284.8 and 293 eV, respectively. In addition to σ* and π* states, as we move from lower to higher energy, three near edge features were observed in the XAS spectrum of graphene oxide at 286.2, 287.8 and 289 eV. These electronic states were attributed to the presence of different functionalities like –C–O, CO and O–CO, respectively. The enhanced intensity with the evolution of new electronic states in the C_K edge spectra of graphene oxide at 287.8 and 289 eV evidently confirm the formation of graphene oxide with a good degree of oxidation. These types of features were not observed in the reference graphite. Therefore, oxidation of graphite and formation of graphene oxide were confirmed. Fig. 6b shows the O_K edge of graphene oxide. The sharp near edge structure of the spectrum of graphene oxide was attributed to the σ* state of OH. However, the small hump near the pre and post edges confirmed the different types of oxygen bindings. Therefore, the comparative study of C and O_K edges of graphene oxide confirmed the formation of graphene oxide with a good degree of oxidation.
 | ||
| Fig. 6 Near edge X-ray absorption fine structure (NEXAFS) (a) comparison of C_K edge of Gt and GrO (b) O_K edge of GrO. | ||
The thermal decomposition of graphene oxide was studied with thermogravimetric analysis (TGA), by monitoring the mass loss of substance as a function of temperature.37 Dynamic nitrogen (industrial grade) at a rate of 100 mL min−1 was used for the measurements, with a slow ramp rate of 10 °C min−1 over 50–600 °C. The thermogravimetric curves of graphene oxide are depicted in Fig. 7. The graphene oxide showed a single stage decomposition TGA curve, indicating rapid mass loss just above room temperature. The significant mass loss for graphene oxide in the DTA spectrum was found at 197.7 °C and total % wt loss was ∼88.51%, due to the removal of thermally labile oxygen-containing groups. The graphene oxide shows a significant weight loss below 200 °C, indicating the decomposition of oxygen-containing functionalities over the surface of graphene oxide.
 | ||
| Fig. 7 Thermal analysis of graphene oxide (a) TGA and (b) DTA. | ||
The TEM measurements of graphene oxide were conducted with JEOL-2100 (Fig. 8).37 The homogeneous dispersions of graphene oxide were prepared in ethanol under sonication for 90 min. 5 μL homogeneous dispersions of graphene oxide were uniformly spread over a 200 mesh holey carbon-coated copper grid. The grid was dried for 6 h at RT, under a closed Petri dish. A low magnification image of graphene oxide was captured to confirm the formation of a graphene oxide sheet (Fig. 8a). The high resolution images reveal the surface morphology of graphene oxide (Fig. 8b). The images confirm the formation of a graphene oxide sheet. The crystalline behavior of graphene oxide has been further confirmed from the corresponding selected area electron diffraction patterns (Fig. 8c). The crystalline behavior of graphene oxide confirmed its ordered arrangement.66,67 The well-resolved concentric rings and spots in the electron diffraction pattern of graphene oxide evidently confirmed the formation of crystalline graphene oxide with a lower number of layers. The six-fold pattern in the diffraction of graphene oxide is consistent with its hexagonal lattice (a = b ≠ c, α = β = 90° and γ = 120°), implying that the graphitic AB stacking order is preserved in the lattice even after a higher degree of oxidation. The dark spots in the first ring of graphene oxide correspond to the ordered graphitic lattice and a relatively low intensity spot in the second ring corresponds to a disordered domain. The d spacing value of the first ring in the SAED pattern of graphene oxide implies that inter-planar distance between the two layers of graphene oxide is 0.20 nm. However, as we move to the second ring the inter-planar distance has reduced to 0.12 nm and the intensity of spots has also been lowered. This verifies the orientation of the functionalities over the surface of graphene oxide. Therefore, it is confirmed that the surface of graphene oxide consists of sp2 and sp3 hybridized carbon atoms.
 | ||
| Fig. 8 HRTEM of GrO (a) low magnification (b) high magnification (c) SAED pattern. | ||
The AFM data were collected in non-contact mode (Fig. 9).36,60,61 The sample was mounted on a glass slide. The dispersions of graphene oxide were prepared with ultrasonication. The homogeneous dispersion of graphene oxide was filtered through 0.45 μm PTFE membranes. The collected filtrate was again sonicated for 90 min. The homogeneously dispersed graphene oxide was mounted over a glass slide and small clumps were cleaned with ethanol. The ethanol was evaporated at RT. The sample was dried with a hair drier. The 6 μm area was scanned to produce a topographical view of the graphene oxide sheet (Fig. 9a). The differences between the two red arrows in the topological view depict the characteristic values of the marked region (Table 1). The marked region between two red arrows has a surface roughness of 10.898 nm (Table 1). The 3-D image of the graphene oxide surface confirmed the surface roughness (Fig. 9b).
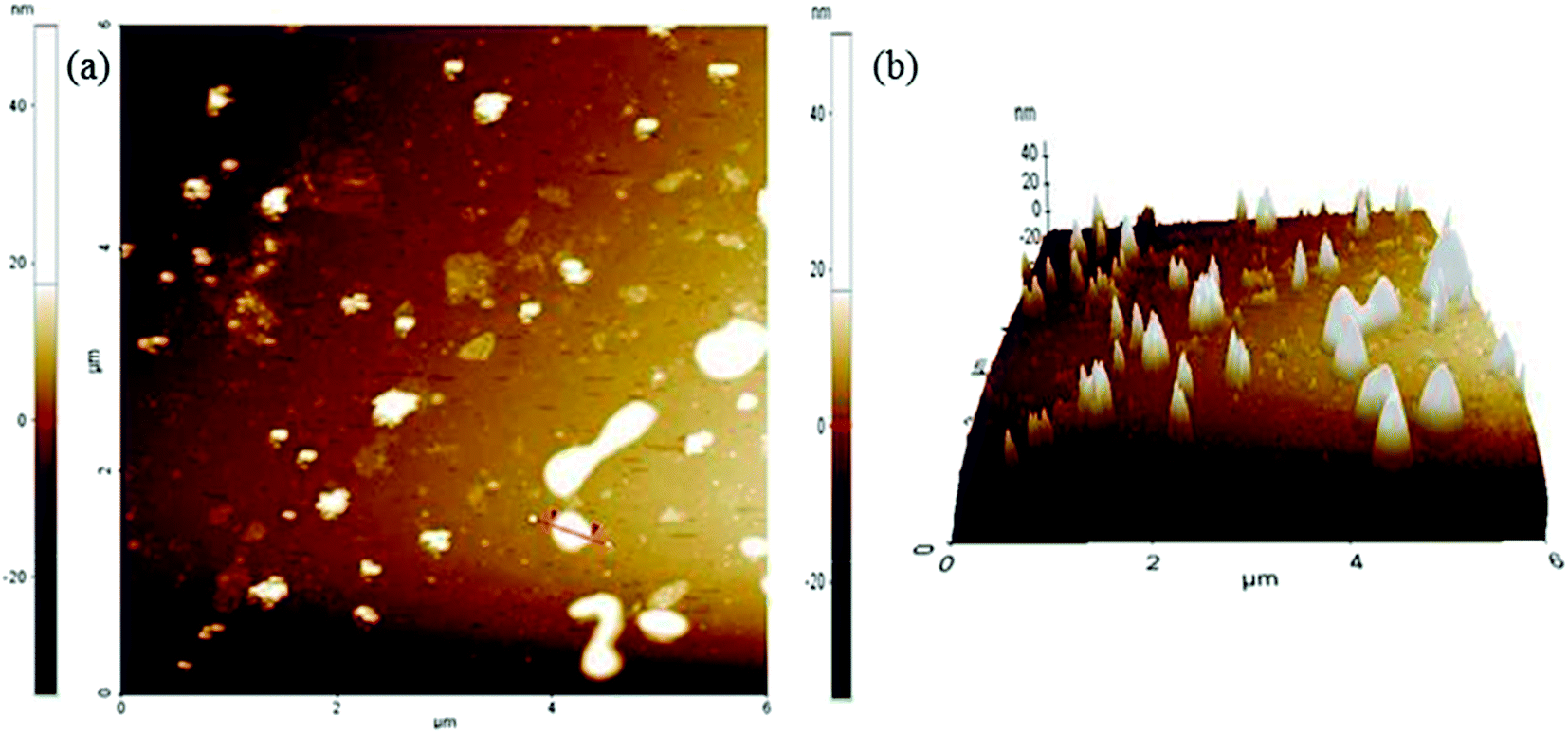 | ||
| Fig. 9 AFM analysis of graphene oxide (a) topological view (b) 3D view. | ||
| Sample | Max (nm) | Mean (nm) | Rpv (nm) | Rq (nm) | Ra (nm) | Rz (nm) |
|---|---|---|---|---|---|---|
| Graphene oxide | 36.211 | 17.669 | 31.098 | 11.803 | 10.898 | N/A |
The morphological investigations were conducted with SEM (Fig. 10).36 The stub used for sample preparation was covered with carbon tape. The powdered samples were adhered on the carbon tape. The stub was exposed for plasma sputtering inside the coater chamber. A target containing gold and palladium in 80:20 ratios was used for the coating. A 20 kV beam voltage was applied to excite the secondary electrons from the surface of the sample. The 20 μm scanning area was chosen to capture the morphological image of graphene oxide. The surface roughness in the morphological images of graphene oxide is in close agreement with the surface roughness data, acquired from AFM. Hence, the morphological characterization indicates the formation of the rough surface of graphene oxide. The results of all structural investigations are in close agreement with each other.
 | ||
| Fig. 10 Morphological analysis of graphene oxide with SEM. | ||
3.2 In vitro cytocompatibility profile
The in vitro cytotoxicity screening was performed on mammalian cell lines, human breast cancer cell line MCF-7 and monkey normal kidney cell line Vero (Fig. 11).‡ The sulforhodamine B (SRB) assay was used for the estimation of cell growth.63,64 In both the experiments four different dose levels 10, 20, 40 and 80 μg mL−1 were used in dimethyl sulfoxide (DMSO). Adriamycin (ADR) was used as a positive control in both experiments. Each experiment was repeated thrice and the mean reading was considered to plot a graph between % control growth and concentration. The GI50 (concentration of drug that produces 50% inhibition of the cells), TGI (concentration of drug that produces the total inhibition of the cells) and LC50 (concentration of the drug that kills 50% of the cells) were calculated from the mean graph. The in vitro experiments on the MCF-7 and Vero cell lines are significant. The graphene oxide was screened against the MCF-7 cell line (Fig. 11a). At four different dose levels of 10, 20, 40 and 80 μg mL−1, the % control growth for graphene oxide against the MCF-7 cell line was 99.39, 97.09, 116.94 and 152.04, respectively. The >80 GI50 value confirmed the low toxicity and excellent cytocompatibility of graphene oxide. Therefore, the low cytotoxicity of graphene oxide has encouraged a probe of its biocompatibility behaviour with normal cell lines. Hence, the studies were continued and the compounds were screened against the normal Vero cell line. Again, exciting results were obtained. The graphene oxide showed an excellent compatibility with Vero cells, as compared to reference ADR (Fig. 11b). At four different dose levels 10, 20, 40 and 80 μg mL−1, the % control growth for graphene oxide against the Vero cell line was 93.9, 108.1, 121.3 and 155.9, respectively. Therefore, graphene oxide was revealed to be a biocompatible material. The morphological effect on the Vero cell lines clearly indicates the biocompatibility of graphene oxide (Fig. 12). Fig. 12a shows the tubular morphology of Vero cells. In Fig. 12b, the tubular morphology of Vero cells was completely damaged after employing the reference ADR. However, Fig. 12c evidently confirmed the least damage to the morphology of Vero cells. The maximum number of cells retain their morphology after addition of graphene oxide. Therefore, the morphological effect evidently confirmed the biocompatibility of graphene oxide. Therefore, graphene oxide could be used as a biomaterial for biocompatible coating. | ||
| Fig. 11 In vitro screening of graphene oxide on (a) MCF-7 and (b) Vero cell lines. | ||
 | ||
| Fig. 12 Digital micrograph of (a) control (b) ADR loaded on control (c) graphene oxide loaded on control. | ||
3.3 Antioxidant activity
The antioxidant activity of graphene oxide was evaluated with 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay (Fig. 13a). A stock solution of DPPH in ethanol was prepared with 2.16 mg/50 mL and the absorbance was recorded at 515 nm.68,69 20, 60, 80, 100 mg mL−1 samples of graphene oxide were prepared to study the antioxidant activity. A 100 mg mL−1 stock solution of L-ascorbic acid was prepared to use as a reference antioxidant. 1.5 mL of sample solution and 1.5 mL of DPPH stock solution were added and incubated in the dark for 45 min. Then the absorbance was measured after 45 min at 515 nm and the percentage scavenging activity was calculated with the following equationwhere AC is the absorbance of DPPH, AS is the absorbance of the test sample. The plot of % scavenging activity of graphene oxide is shown in Fig. 13b. The % antioxidant activity of graphene oxide is found in the range 50.57–45.89%. A linear decrease in antioxidant capacity with increase in concentration was observed for graphene oxide. The antioxidant activity of graphene oxide was complemented by the findings of in vitro cellular interactions. Therefore, graphene oxide is found to be significant for biological applications.

 | ||
| Fig. 13 Antioxidant activity of graphene oxide (a) DPPH assay (b) % scavenging activity. | ||
3.4 In vitro antimicrobial assay
The functionalization of graphene has enhanced the suitability of graphene for biological applications. Therefore, the surface modification of graphene has emerged as a new technique for biomedical applications. Usually, nitrogen and sulphur doped compounds are well known for antimicrobial assay.60 However, in the present studies, the significance of oxygen doped graphene has been demonstrated with detailed mechanistic insights towards antimicrobial activity. The various states of oxygen doped onto a graphenic framework have enhanced the biological efficacy of graphene. The synergistic impact of contributory factors (SICF) has revealed the mechanism of antimicrobial activity. The antimicrobial experiments on graphene oxide were conducted with NCIM strains like Gram-positive bacteria, S. aureus, B. subtilis and Gram-negative bacteria, E. coli, P. aeruginosa. However, antifungal activity is evaluated with C. albicans and A. niger. Surprisingly, we never obtained any activity against all the above mentioned strains. The strange behaviour of graphene oxide with NCIM strains is scientifically ascribed to increased genetic variability during the sub-culturing of strains. The results of these findings are demonstrated in Table 2. However, the highly reproducible ATCC strains have confirmed the antimicrobial activity of graphene oxide. The antimicrobial evaluation of graphene oxide has led by the above experimental assay to an antimicrobial test (Table 3). This assay has confirmed the significance of graphene oxide for biological systems.65 The antibacterial activity of graphene oxide was perceived with ATCC strains, against human pathogenic Gram-positive S. aureus and Gram-negative E. coli by the tube dilution method using DMSO and DMF solvents on a Mueller Hinton broth. Similarly, graphene oxide was also screened against antifungal agent C. albicans. The obtained results are very interesting. The outstanding minimum inhibitory concentration (MIC) of graphene oxide was perceived against antifungal agent at 128 mg mL−1. The MIC against antibacterial strains, S. aureus and E. coli was obtained at 256 mg mL−1. Hence, graphene oxide has emerged as an advanced functional material for antimicrobial coatings.| Sample | S. aureus NCIM 2079 | B. subtilis NCIM 2250 | P. aeruginosa NCIM 2036 | E. coli NCIM 2109 | C. albicans NCIM 3471 | A. niger NCIM 545 |
|---|---|---|---|---|---|---|
| Graphene oxide | Resistant | Resistant | Resistant | Resistant | Resistant | Resistant |
| Graphene oxide | S. aureus ATCC 25923 | E. coli ATCC 25922 | C. albicans ATCC 10231 |
|---|---|---|---|
| MIC | 256 mg mL−1 | 256 mg mL−1 | 128 mg mL−1 |
3.5 Synergistic impact of contributory factors (SICF)
The significance of graphene oxide for biomedical applications has been confirmed through extensive analytical techniques. 13C CP-MAS SSNMR confirmed different types of carbons with oxygen functionalities. The results are in close agreement with HRXPS. Through surface interactions, different oxygen functionalities generate reactive functional interfaces with sp2 domains. Therefore, the contribution of various functionalities has been confirmed with SR-NEXAFS, 13C CP-MAS SSNMR, XRD, HRXPS, HRTEM, SAED, AFM, SEM, Raman, TGA, DTA, FTIR, UV, BET and a Porosimeter. Furthermore, in vitro biomedical applications of graphene oxide, like antimicrobial activity, cytotoxicity and antioxidant efficacy, have endorsed the contribution of the functional interface. Owing to its lower energy, the graphite has higher stability and hence least processability in organic solvents. Also, the higher stability of graphitic carbon is attributed to its ordered sp2 domains. The ordered sp2 domains in graphite have been reduced after surface functionalization. Fig. 2a has confirmed the presence of a D peak at 1359 cm−1, corresponding to defective crystal lattice. These defects have arisen due to surface functionalization. The enhanced defects over the surface of graphene oxide impart higher functional stability as compared to graphite. Therefore, the surplus oxygen-containing functionalities in graphene oxide have confirmed its suitability for various biomedical applications. Hence, the formation of a functional interface enhanced disorder to render functional stability to a defective crystal lattice.27 Hence, together with the functional interface, the other influential factors are• Holy sp2 domains render active centers for surface functionalization with featured edges.
• Functional atomic domains create reactive interfaces for biomedical applications.
• The multifunctional surface has actively contributed towards cellular interactions, particularly oxygen species.
• The surface area and morphology, roughness, pore shape and morphology have synergistically contributed towards biological activity.
Therefore, the synergistic impact of all the contributory factors has confirmed the suitability of graphene oxide for biomedical applications. Owing to the SICF mechanism, the cytocompatibility of graphene oxide has been confirmed on MCF-7 and Vero cell lines. The SRB assay has confirmed the higher cell viabilities, >80% for graphene oxide. The enhanced cell viabilities as compared to standard ADR have confirmed the suitability of graphene oxide towards biocompatible material. The ability of a compound for free radical scavenging is noted as an antioxidant activity. The highly reactive surplus oxygen-containing species over the surface of graphene oxide have considerable significance for antioxidant behaviour. The DPPH assay has confirmed the significance of graphene oxide for antioxidant activity. The antimicrobial assay has confirmed the significance of graphene oxide for antimicrobial activity. The lower MIC values for antifungal as compared to antibacterial strains have confirmed the suitability of graphene oxide for antifungal coatings. However, significant antibacterial activity was perceived against both bacterial strains. Therefore, the surplus oxygen-containing functionalities facilitate the biomedical applications of graphene oxide (Scheme 1).
 | ||
| Scheme 1 Synthesis of graphene oxide. | ||
4. Conclusions
The multifunctional surface of graphene oxide has been confirmed with extensive structural investigations. The functional atomic domains with a defective crystal lattice have been confirmed with Raman spectroscopy. The contribution of edge structures with evolved electronic transitions has been confirmed with synchrotron radiation near edge X-ray absorption fine structure (SR-NEXAFS). The surplus oxygen-containing functionalities have reduced the toxicity of graphene. Therefore, the impact of oxygen-containing functionalities has significance for biomedical applications of graphene oxide. Hence, graphene oxide has emerged as an advanced functional material and the proposed mechanism of synergistic impact of contributory functionalities (SICF) is feasible for biomedical applications of graphene-based materials.Acknowledgements
Authors are thankful to Central University of Gujarat for infrastructural support and instrumentation facility. UGC, New Delhi is acknowledged for fellowship support. The National Chemical Laboratory is acknowledged for the solid-state NMR facility. Dr Dinesh Kumar Shukla and Mr Rakesh Kumar Sah, UGC-DAE CSR Indore, were acknowledged for their valuable guidance and help for soft X-ray absorption measurements, respectively. Dr Vasant Sathe, UGC-DAE CSR Indore, was acknowledged for providing Raman facility. Dr Ulhas Patil, Microbiology Department, R. C. Patel College, Shirpur, Maharashtra, was acknowledged for antimicrobial tests and fruitful discussion. Dr Jyoti A. Kode, ACTREC, Tata Memorial Centre, Mumbai, was acknowledged for providing in vitro cytotoxicity screening facility.References
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Z. Jiang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- M. I. Katsnelson, Mater. Today, 2007, 10, 1–2 CrossRef.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- D. Ayrat, V. K. Dmitry, B. A. Lawrence, C. Pavel and J. M. Tour, J. Am. Chem. Soc., 2012, 134, 2815–2822 CrossRef PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- A. Clauss, R. Plass, H. P. Boehm and U. Hofmann, Z. Anorg. Allg. Chem., 1957, 291, 205–220 CrossRef CAS.
- W. Scholz and H. P. Boehm, Z. Anorg. Allg. Chem., 1969, 369, 327–340 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- J. Gu, N. Li, L. Tian, Z. Lv and Q. Zhang, RSC Adv., 2015, 5, 36334 RSC.
- J. Gu, X. Yang, Z. Lv, N. Li, C. Liang and Q. Zhang, Int. J. Heat Mass Transfer, 2016, 92, 15 CrossRef CAS.
- J. Gu, X. Yang, C. Li and K. Kou, Ind. Eng. Chem. Res., 2016, 55, 10941 CrossRef CAS.
- J. Gu, C. Liang, J. Dang, W. Dong and Q. Zhang, RSC Adv., 2016, 6, 35809 RSC.
- H. Gu, C. Ma, J. Gu, J. Guo, X. Yan, J. Huang, Q. Zhang and Z. Guo, J. Mater. Chem. C, 2016, 4, 5890 RSC.
- W. Zhao, J. Kong, H. Liu, Q. Zhuang, J. Gu and Z. Guo, Nanoscale, 2016 10.1039/C6NR06622D.
- L. Zhang, Q. Zhang, H. Xiea, J. Guoc, H. Lyu, Y. Li, Z. Sun, H. Wang and Z. Guo, Appl. Catal., B, 2017, 201, 470–478 CrossRef CAS.
- B. Jiang, L. Zhao, J. Guo, X. Yan, D. Ding, C. Zhu, Y. Huang and Z. Guo, J. Nanopart. Res., 2016, 18, 145 CrossRef.
- Q. Zhang, D. Jiang, L. LiU, Y. Huang, J. Long, G. Wu, Z. Wu, A. Umar, J. Guo, X. Zhang and Z. Guo, J. Nanosci. Nanotechnol., 2015, 15, 9807–9811 CrossRef CAS PubMed.
- K. Wang, J. Ruan, H. Song, J. Zhang, Y. Wo, S. Guo and D. Cui, Nanoscale Res. Lett., 2011, 6, 8 Search PubMed.
- Y. Liu, D. Yu, C. Zeng, Z. Miao and L. Dai, Langmuir, 2010, 26, 6158–6160 CrossRef CAS PubMed.
- L. Feng and Z. Liu, Nanomedicine, 2011, 6, 317–324 CrossRef CAS PubMed.
- P. Zuo, H. Feng, Z. Xu, L. Zhang, Y. Zhang, W. Xia and W. Zhang, Chem. Cent. J., 2013, 7, 39 CrossRef PubMed.
- S. A. Sydlik, S. Jhunjhunwala, M. J. Webber, D. G. Anderson and R. Langer, ACS Nano, 2015, 9, 3866–3874 CrossRef CAS PubMed.
- A. M. Pinto, C. Gonçalves, D. M. Sousa, A. R. Ferreira, J. A. Moreira, I. C. Gonçalves and F. D. Magalhaes, Carbon, 2016, 99, 318–329 CrossRef CAS.
- K. Urbas, M. Aleksandrzak, M. Jedrzejczak, M. Jedrzejczak, R. Rakoczy, X. Chen and E. Mijowska, Nanoscale Res. Lett., 2014, 9, 656 CrossRef PubMed.
- J. Wang, Y. Wei, X. Shi and H. Gao, RSC Adv., 2013, 3, 15776–15782 RSC.
- S. Eigler, M. E. Heim, S. Grimm, P. Hofmann, W. Kroener, A. Geworski, C. Dotzer, M. Rockert, J. Xiao, C. Papp, O. Lytken, H. P. Steinruck, P. Muller and A. Hirsch, Adv. Mater., 2013, 25, 3583–3587 CrossRef CAS PubMed.
- S. Stankovich, D. Dikin, R. Piner, K. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. Nguyen and R. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- Z. Sun, D. K. James and J. M. Tour, J. Phys. Chem. Lett., 2011, 2, 2425–2432 CrossRef CAS.
- R. S. Dey, S. Hajra, R. K. Sahu, R. Raj and M. K. Panigrahi, Chem. Commun., 2012, 48, 1787–1789 RSC.
- S. Gurunathan, J. W. Han and J. H. Kim, Int. J. Nanomed., 2013, 8, 2719–2732 CrossRef PubMed.
- S. S. Maktedar, S. S. Mehetre, M. Singh and R. K. Kale, Ultrason. Sonochem., 2014, 21, 1407–1416 CrossRef CAS PubMed.
- S. S. Mehetre, S. S. Maktedar and M. Singh, Appl. Surf. Sci., 2016, 366, 514–522 CrossRef CAS.
- H. Zhang, G. Gruner and Y. Zhao, J. Mater. Chem. B, 2013, 1, 2542–2567 RSC.
- R. Zappacosta, M. Di Giulio, V. Ettorre, D. Bosco, C. Hadad, G. Siani, S. Di Bartolomeo, A. Cataldi, L. Cellini and A. Fontana, J. Mater. Chem. B, 2015, 3, 6520–6527 RSC.
- S. Sayyar, E. Murray, B. C. Thompson, J. Chung, D. L. Officer, S. Gambhir, G. M. Spinks and G. G. Wallace, J. Mater. Chem. B, 2015, 3, 481–490 RSC.
- P. Gandhiraman, D. Nordlund, C. Javier, J. E. Koehne, B. Chen and M. Meyyappan, J. Phys. Chem. C, 2014, 118, 18706–18712 Search PubMed.
- D. Li, M. Muller, S. Gilje, R. Kaner and G. Wallance, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25–29 CrossRef CAS PubMed.
- B. Brodie, Philos. Trans. R. Soc. London, 1859, 14, 249–259 CrossRef.
- U. Hofmann and R. Holst, Ber. Dtsch. Chem. Ges. A, 1939, 72, 754–771 CrossRef.
- G. L. Ruess, Monatsh. Chem., 1947, 76, 381–417 CrossRef CAS.
- T. Niakajima, A. Mabuchi and R. Hagiwara, Carbon, 1988, 26, 357–361 CrossRef.
- M. L. Bhaisare, B. S. Wu, M. C. Wu, M. S. Khan, M. H. Tseng and H. F. Wu, J. Biomater. Sci., Polym. Ed., 2016, 4, 183–194 RSC.
- S. S. Banerjee, A. Badhwar, P. Wate, S. Asai, K. R. Zope, R. Mascarenhas, D. Bhatia and J. Khandare, J. Biomater. Sci., Polym. Ed., 2014, 2, 57–66 RSC.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- M. Mermoux, Y. Chabre and A. Rousseau, Carbon, 1991, 29, 469–474 CrossRef CAS.
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef CAS.
- W. Gao, L. B. Alemany, C. Lijie and P. M. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS PubMed.
- H. K. Jeong, Y. P. Lee, R. J. W. E. Lahaye, M. H. Park, K. H. An and I. J. Kim, J. Am. Chem. Soc., 2008, 130, 1362–1366 CrossRef CAS PubMed.
- X. Li, Q. Zhang and L. Chen, Chin. J. Chem., 2010, 68, 1915–1920 CAS.
- M. Nurunnabi, K. Parvez, M. Nafiujjaman, V. Revuri, H. A. Khan, X. Feng and Y. K. Lee, RSC Adv., 2015, 5, 42141–42161 RSC.
- W. Zhang, L. Wu, Z. Li and Y. Liu, RSC Adv., 2015, 5, 49521–49533 RSC.
- E. L. K. Chng and M. Pumera, RSC Adv., 2015, 5, 3074–3080 RSC.
- B. Li, Y. Nan, P. Zhang and X. Song, RSC Adv., 2016, 6, 19797–19806 RSC.
- S. S. Maktedar, S. S. Mehetre, G. Avashthi and M. Singh, Ultrason. Sonochem., 2017, 34, 67–77 CrossRef CAS PubMed.
- S. S. Maktedar, G. Avashthi and M. Singh, Ultrason. Sonochem., 2017, 34, 856–864 CrossRef CAS PubMed.
- J. Wang, X. Mei, L. Huang, Q. Zheng, Y. Qiao, K. Zheng, S. Mao, R. Yang, Z. Zhang, Y. Gao, Z. Guo, Z. Huang and Q. Wang, J. Energy Chem., 2015, 24, 127–137 CrossRef.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- P. Skehn, R. Storeng, A. Scudiero, J. Monks, D. McMohan, D. Vistica, T. W. Jonathan, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107 CrossRef.
- I. Barbolina, C. R. Woods, N. Lozano, K. Kostarelos, K. S. Novoselov and I. S. Roberts, 2D Mater., 2016, 3, 025025 CrossRef.
- M. Patange, S. Biswas, A. K. Yadav, S. N. Jha and D. Bhattacharyya, Phys. Chem. Chem. Phys., 2015, 15, 32398–32412 RSC.
- G. K. Inwati, Y. Rao and M. Singh, Nanoscale Res. Lett., 2016, 11, 458 CrossRef PubMed.
- P. Malik, R. K. Ameta and M. Singh, Chem.-Biol. Interact., 2014, 222, 77–86 CrossRef CAS PubMed.
- P. Malik, R. K. Ameta and M. Singh, J. Mol. Liq., 2016, 220, 604–622 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23416j |
| ‡ Source of in vitro cytocompatibility study: the in vitro results were obtained on mammalian cell lines, human breast cancer cell line MCF-7 and monkey normal kidney cell line Vero. |
| This journal is © The Royal Society of Chemistry 2016 |