
Decoration of GO with Fe spinel-Naf/DMAP: an electrochemical probe for sensing H2O2 reduction†
Abstract
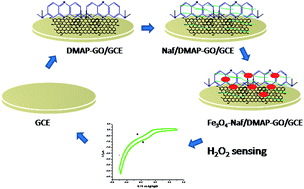
We herein report the preparation of graphene oxide decorated with Fe spinel (Fe3O4)-Naf/DMAP for an unprecedented and highly selective non-enzymatic electrochemical sensing of hydrogen peroxide reduction. The linking of 3,7-bis (dimethylamino)-phenothiazin-5-ium chloride (DMAP) to the graphene oxide occurred via electrostatic interactions of the cationic organic compound with negatively charged oxygen-containing groups (–COO− and –O−) available on the edges of the graphene oxide. Fe3O4 (Fe2+, Fe3+) nanoparticles were adhered further on SO3− moieties of the Nafion coated over the DMAP-GO/GCE through electrostatic interactions. This green approach was used to prepare a derived graphene oxide hybrid nano-material. This material was characterized using scanning electron microscopy (SEM), transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FTIR), UV-visible spectroscopy, X-ray diffraction (XRD), and cyclic voltammetry (CV). The prepared material was utilized to detect H2O2 subjected to electrocatalytic reduction and to determine its concentration in the linear range of 5.5 μM to 2.4 mM. Nafion was employed as a dispersion medium, and an anchor for Fe3O4 to make the composite and to prevent interfering effects of other metabolites. The detection limit of this highly selective and highly reproducible film was estimated to be 0.6 μM with a sensitivity of 32.0 μA mM−1 cm−2.
 Please wait while we load your content...
Please wait while we load your content...

