
Structural and thermal analysis of a hyper-branched exopolysaccharide produced by submerged fermentation of mushroom mycelium†
L. Chen ac,
W. N. Chengb,
B. B. Zhanga and
P. C. K. Cheung*c
ac,
W. N. Chengb,
B. B. Zhanga and
P. C. K. Cheung*c
aKey Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi 214122, China
bDepartment of Pharmacy, Affiliated Hospital of Binzhou Medical University, Binzhou 256603, China
cSchool of Life Sciences, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, China. E-mail: petercheung@cuhk.edu.hk
First published on 22nd November 2016
Abstract
An exopolysaccharide (PTR-EPS1) purified from the culture medium of the mycelial fermentation of Pleurotus tuber-regium had a weight-average molecular mass of 173.6 kDa and a radius of 55.6 nm. Methylation analysis and NMR results indicated that PTR-EPS1 had a 81% degree of branching at O-2 and consisted of a backbone of 1,6-linked α-D-Manp residues branched at O-2 with either a terminal α-D-Manp or -(1,2)-α-D-Manp-(1,2)-α-D-Manp units. The morphology of PTR-EPS1 was found to be spherical by TEM. Thermal analysis indicated that the main thermal degradation (up to 77.56% dry mass) of PTR-EPS1 occurred within a temperature range of 205.7–331.2 °C. The apparent activation energy Ea and pre-exponential factor A of PTR-RES1 were found to be 223.4 kJ mol−1 and 5.37 × 1023 s−1, respectively. This is the first report on the structural information and thermal degradation properties of PTR-EPS1. These results will facilitate the potential application of this mushroom polysaccharide as a novel biopolymer in the design of carbohydrate-based nanoparticles and food delivery systems.
Introduction
Highly branched macromolecules have attracted increasing attention in the fields of nanotechnology and pharmacology because of their unique spherical structures which provide multiple terminal units to which different functional groups can be attached to produce novel nanomaterials.1,2 Compared with synthetic highly branched polymers, research interest in natural hyper-branched polysaccharides (HBPs) is emerging in the field of food bioactive compounds delivery systems due to their non-toxicity, good biocompatibility and biodegradability.3 Nanoparticles formed by polysaccharides can deliver a variety of food bioactive components (including antioxidants, probiotics, polyunsaturated fatty acids, and proteins) to improve their stability, solubility, cellular uptake, bioavailability and may also provide controlled release for better efficacy.4,5 Spherical HBPs do not only provide reaction sites for the formation of nanoparticles, but they also protect the nanoparticles in a shell structure with excellent dispersion in water.3,6 These unique properties of HBPs can be applied in the fields of food delivery and controlled release.6 Recent studies on HBPs have been focused on obtaining them from natural sources and characterizing their structure and physical properties including solubility, shrinking factors, and rheological properties.7,8 Some natural biological macromolecules such as chitosan, xylan and starch have been studied for their application in nanoparticles for drug delivery.9–11 However, there is very little study on the morphology and thermal degradation behavior of HBPs, which would greatly affect their potential applications as novel biomaterials in food components and drug delivery systems.Mushroom has been considered to be a good source of HBPs.12 Recently, a mushroom HBP was isolated from the sclerotium of Pleurotus tuber-regium (PTR) with structural features that have potential application as nanomaterial in food/drug delivery and gene vector.13 Another mushroom HBP which was a highly branched (62.5% degree of branching) exopolysaccharide (EPS) was also obtained from the culture medium of submerged fermentation of PTR mycelium.14 The production of this EPS could be enhanced by the addition of Tween 80 into the medium and its mechanism was also studied by proteomic approach.15,16 However, the pyrolysis properties of these HBPs, which would greatly affect their further application, have not been investigated previously.
Isoconversional thermal analysis method has been employed as a powerful technique to monitor physical and chemical changes in both natural and synthetic polymers for their applications as biopolymers in food industry.17,18 In particular, it can provide essential information regarding thermal decomposition kinetics of drug delivery system, e.g. β-cyclodextrin inclusion complex.19,20 Parameters for thermal decomposition kinetics such as apparent activation energy (Ea) are commonly analyzed by both the model-free and model-fitting approaches. In model-free methods, Ea is determined based on the experimental data without any assumptions about the theoretical reaction function and reaction order. Thus it eliminates the limitations of other methods such as ASTM E1641 and E698 methods which assume that the Ea remains constant throughout the degradation step.17,21 In contrast, model-fitting methods allow one to make an averaging of Ea for different steps of decomposition, whereas the number of steps in question should be established by the model-free methods.22
In the present work, a homoglycan type hyper-branched EPS (PTR-EPS1) was purified from the submerged fermentation medium of PTR mycelium and its fine structure and molecular conformation were characterized. Furthermore, the isoconversional thermal analysis was used to determine the thermal stability, moisture content and thermal degradation pattern of this polysaccharide by model-free and model-fitting approaches. The structural information and thermal properties can facilitate further application of this biopolymer in food delivery system.
Materials and methods
Culture conditions and yield of EPS
The mycelium of Pleurotus tuber-regium (PTR) was purchased from Fungi Perfecti, Ltd. Co (Olympia, USA). EPS was isolated from the culture medium of PTR mycelium in submerged fermentation using shake flasks as described in our previous studies.14 After centrifugation (4000g for 10 min) and filtration, EPS was isolated by fractional ethanol precipitation from the mycelium-free culture medium. Initially, 1/3 volume of 95% ethanol was added to remove the macromolecules and this was followed by three volumes of 95% ethanol to precipitate the target EPS. The crude EPS was obtained by filtration and was re-dissolved in double distilled water (DD H2O) and precipitated by three volumes of 95% ethanol repeatedly for three times. The precipitated EPS were then separated by centrifuging at 4000g for 15 min. The yield of EPS was determined gravimetrically after lyophilization by use of a freeze dryer at −70 °C under vacuum.Purification of EPS
The above EPS fraction was then purified by ultrafiltration membrane with 10 kDa molecular weight cut-off to remove any small molecules present. The retained EPS was subjected to gel filtration chromatography using a Sephacryl S-400 High-Resolution column (90 × 2.6 cm i.d., Amersham Pharmacia Biotech, UK) eluted with DD H2O at 1.5 mL min−1. The eluted fraction was then freeze dried to give the purified EPS fraction (PTR-EPS1).Chemical composition analysis
Total proteins, neutral sugars and uronic acid content of the PTR-EPS1 were determined using bicinchoninic acid (BCA) method,23 phenol-sulfuric acid method24 and the modified meta-hydroxy diphenyl-sulfuric acid method,25 respectively. Monosaccharide composition of PTR-EPS1 was determined by the alditol acetate sugar derivatives prepared by acid hydrolysis, reduction, and acetylation, followed by the quantification using GC-MS (6890N-5973N, Agilent Technology, USA) with an Alltech DB-225 capillary column (15 m × 0.25 mm i.d., 0.25 μm film) as described previously.26 The oven temperature were heated from 170 °C to 220 °C at 2 °C min−1 with a final hold for 15 min. Helium was used as the carrier gas at a flow rate of 1.0 mL min−1. The injector temperature and interface temperature were set at 250 °C and 280 °C, respectively. The MS conditions were set as follows: the ion source temperature at 250 °C, ionization energy at 70 eV, detector voltage at 1.5 kV, and mass range from 50 to 400.Structural elucidation of PTR-EPS1
The weight-averaged molecular weight (Mw) was determined from Debye plot based on the Rayleigh equation (eqn (1)) with K, Mw, A2, and C stand for optical constant, sample molecular weight, 2nd virial coefficient, and concentration, respectively.
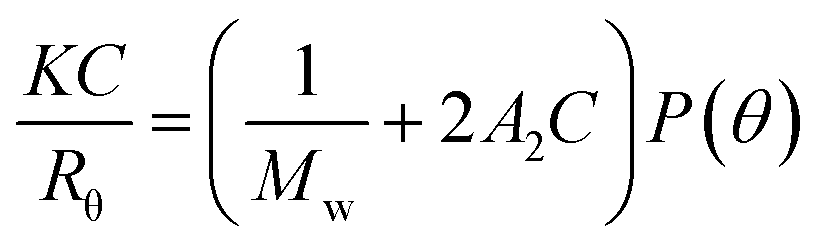 | (1) |
The Rayleigh ratio (Rθ) indicates the ratio of scattered light to incident light of the sample, while P(θ) means the angular dependence of the sample scattering intensity. When the particles in solution are much smaller than the wavelength of the incident light, multiple photon scattering could be avoided. Under these conditions, P(θ) would reduce to 1 and the angular dependence of the scattering intensity was lost that resulted in the Rayleigh scattering. The Mw, expressed in Daltons (or g mol−1), was then determined from the intercept at zero concentration (C → 0) according to KC/Rθ = 1/Mw.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w) and heated together at 80 °C for 2 h. The EPS control and SDS solution of PTR-EPS1 were diluted stepwise to 100, 50, 10 and 5 μg mL−1, followed by heating to 80 °C for 2 h with constant stirring. After filtration through a 0.22 μm nylon syringe filter, a droplet of the sample (5 μg mL−1) was deposited on the holey carbon film specimen (200 mesh, Beijing Zhongjingkeyi Technology, China), which was finally dried in air at ambient temperature and humidity. Molecular morphology of the prepared samples was visualized by a Transmission electron microscope (TEM, H-7650, Hitachi, Japan) with an accelerating voltage of 80 kV.
:1 (w/w) and heated together at 80 °C for 2 h. The EPS control and SDS solution of PTR-EPS1 were diluted stepwise to 100, 50, 10 and 5 μg mL−1, followed by heating to 80 °C for 2 h with constant stirring. After filtration through a 0.22 μm nylon syringe filter, a droplet of the sample (5 μg mL−1) was deposited on the holey carbon film specimen (200 mesh, Beijing Zhongjingkeyi Technology, China), which was finally dried in air at ambient temperature and humidity. Molecular morphology of the prepared samples was visualized by a Transmission electron microscope (TEM, H-7650, Hitachi, Japan) with an accelerating voltage of 80 kV.Isoconversional thermal analysis
The thermodynamic parameters of PTR-EPS1 were determined by use of a simultaneous thermal analyzer STA (Q-600, TA instruments, USA) containing both the thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC). About 5 mg of PTR-EPS1 were analyzed under nitrogen atmosphere at a flow rate of 100 cm3 min−1. The data were obtained at three different heating rates, including 5, 10 and 20 °C min−1 from 25 to 600 °C, and analyzed by both the model-free and model-fitting approaches.The obtained pyrolysis data were firstly analyzed by the isoconversional Flynn–Wall–Ozawa (FWO) method (eqn (2)) as described previously.30,31
 | (2) |
 ; β the heating rate; A the pre-exponential factor; R the general gas constant, Ea the apparent activation energy and T the temperature at the conversion α. The FWO method was considered to be a model-free approach, which can estimate −Ea/R from the slope of a plot of logβ vs. 1/T at different conversion rates. Additionally, twelve functional forms [g(α)] were selected to identify the best degradation model by a model-fitting approach using Coats–Redfern eqn (3) with T′ being the average experimental temperature.
; β the heating rate; A the pre-exponential factor; R the general gas constant, Ea the apparent activation energy and T the temperature at the conversion α. The FWO method was considered to be a model-free approach, which can estimate −Ea/R from the slope of a plot of logβ vs. 1/T at different conversion rates. Additionally, twelve functional forms [g(α)] were selected to identify the best degradation model by a model-fitting approach using Coats–Redfern eqn (3) with T′ being the average experimental temperature.
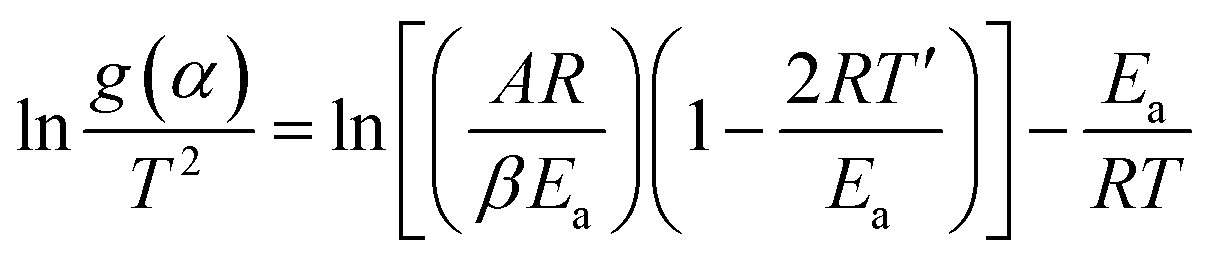 | (3) |
A set of thermal parameters (A and Ea) was yielded by a plot of ln(g(α)/T2) vs. 1/T. The pre-exponential factor A was calculated by use of the compensation effect relationship as eqn (4) where a and b are the compensation parameters. Based on the experimental Ea and A, the reaction rate (k) was then calculated according to the Arrhenius equation (eqn (5)). Then, activation enthalpy (ΔH≠), activation entropy (ΔS≠) and activation free energy (ΔG≠) were obtained according to the thermodynamic eqn (6)–(8).
|
lnA = aEa + b
| (4) |
 | (5) |
 | (6) |
 | (7) |
| ΔG≠ = ΔH≠ − T × ΔS≠ | (8) |
The data were analyzed by the use of Universal Analysis 2000 software (version 4.5A, TA Instruments, USA) and MS Excel 2013.
Results and discussion
Preparation, purification and composition analysis of EPS
In the present study, a fraction with very large molecular weight was removed by adding 1/3 volume of 95% ethanol at the beginning and a second fraction (the target EPS) was obtained when three volumes of 95% ethanol were added. The target crude EPS fraction from PTR mycelium (PTR-EPS) was obtained by lyophilization to give a yield of 0.44 ± 0.07 g L−1. The phenol-sulfuric acid test showed that the total carbohydrate content of PTR-EPS was 97.64 ± 1.47% (wt%), while the negative results of BCA and meta-hydroxydiphenyl-sulphuric acid tests indicated that PTR-EPS did not contain proteins and uronic acids. After purification of PTR-EPS by gel permeation chromatography, a single fraction was obtained and was designated as PTR-EPS1. We have previously optimized the EPS production in submerged fermentation of PTR mycelium by addition of Tween 80.14 The optimal yield of mycelial biomass and EPS production were increased by 51.3% (10.38 g L−1) and 41.8% (1.03 g L−1), respectively, by adding 3.0 g L−1 Tween 80 to the culture medium. However, the EPS produced under such optimized condition was found to be comprised of glucose and mannose in the ratio of 43.4:56.6 with a molecular weight of 3.18 × 106 g mol−1,14 which was in contrast to the present PTR-EPS1 that was only composed of mannose and had a molecular weight of 1.736 × 105 g mol−1.
Structure elucidation of PTR-EPS1
PMAA analysis using GC-MS indicated that PTR-EPS1 consisted of non-reducing terminal mannose (T-Manp), 1,2-Manp and 1,2,6-Manp in a molar ratio of 2.3:1.0:2.1 (Table 1). The ratio between terminal units (T-D-Manp) and branching points (1,2,6-D-Manp) was 1.1 (NT/NB ratio), which indicated that the number of terminal units was approximately equal to the number of branching points in the polysaccharide (Table 1). Results from methylation analysis suggested that the possible partial structure of PTR-EPS1 could be either having →1)-D-Manp-(6→ residues as the backbone with some →1)-D-Manp-(2→ residues at O-2 site to form the side chains or having →1)-D-Manp-(2→ residues as the backbone with some T-D-Manp residues at O-6 site to form the side chains. The degree of branching (DB) value of PTR-EPS1 was calculated to be 81% by the equation [DB = (NT + NB)/(NT + NB + NL)] as shown in Table 1. With such a high DB value, PTE-EPS1 was identified as a hyper-branched mannan. In our previous study, the EPS obtained by adding four volumes of 95% ethanol to PTR mycelium-free culture medium was a hyper-branched glucomannan having a main chain consisted of 1, 6-linked Manp with two [→6)-Manp-(2→] residues and a terminal Glcp at O-2 sites as side chains.14
| Linkage pattern | PMAA | Peak area (%) | Molar ratio |
|---|---|---|---|
| a Data are mean of triplicate.b Manp, Mannopyranose.c NT, NB, and NL are the numbers of the terminal residues, branch residues, and linear residues, respectively. | |||
| T-D-Manpb (NT)c | 1,5-O-Ac2-2,3,4,6-O-Me4-D-mannitol | 39.7 | 2.3 |
| 1,2-D-Manp (NL)c | 1,2,5-O-Ac3-3,4,6-O-Me3-D-mannitol | 18.5 | 1.0 |
| 1,2,6-D-Manp (NB)c | 1,2,5,6-O-Ac4-3,4-O-Me2-D-mannitol | 41.0 | 2.1 |
 | ||
| Fig. 1 1H NMR (a) and 13C DEPT-135 (b) spectra of PTR-EPS1. Residues A, B, C, D, E were designated according to the decreasing order of their chemical shifts. | ||
 | ||
| Fig. 2 The possible partial structure of the repeating units in PTR-EPS1 (a) linkage diagram and (b) space filling model of PTR-EPS1 simulated by ChemBioOffice Ultra 12.0 software, red color represents O atom, grey color represents C atom, and white color represents H atom. | ||
 | ||
| Fig. 3 (a) TEM image of PTR-EPS1 in water showing the presence of fractal dimension structure; (b) extended spherical conformation of EPS molecules dispersed in SDS solution. | ||
However, the spherical conformation of PTR-EPS1 was observed when SDS was added to reduce molecular aggregation (Fig. 3b). According to our previous study,13 SDS could break the hydrogen bonds inside the sphere, but no branched or chain conformation was visualized under this condition. This conformation might be due to the unique chemical structure of PTR-EPS1 that had a high DB value of 81% and a backbone formed by →1)-D-Manp-(6→ linkages with some →1)-D-Manp-(2→ residues at the O-2 position to form the side chains. As shown in Fig. 2b, a computer-simulated partial 3D structure of PTR-EPS1 molecule was shown to be self-winding. Moreover, the average radius of PTR-EPS1 molecule in SDS was found to be 514 ± 29 nm (n = 50), which was almost 10 times of that determined by SLS (55.6 ± 4.3 nm). This result implied that the fractal structure of PTR-EPS1 was opened up by the action of SDS. Moreover, this fractal structure was not the morphology of a single molecule, but a large aggregate formed by a number of small particles.
FD has a wide variety of biomedical applications including biopharmaceutics, pharmacokinetic, as well as advanced Drug Delivery nanoSystems (aDDnSs).34 In a recent study, fractal analysis was applied to quantify the morphological image of the aDDnSs and predict the stability of delivery nanoparticles.35 A previous study on a mushroom polysaccharide (pachyman) using FD theory indicated that its intermolecular hydrogen bonds would lead to aggregation of the polymers.36 Due to the aggregating nature of HBPs, FD might perform an important role in the further development of HBPs as nanoparticles in both food delivery systems and gene vector.
Thermal decomposition characteristics of PTR-EPS1
Thermal decomposition parameters of PTR-EPS1 was determined from the TG, DTG and D2TG (second-time derivatives) curves in the temperature range of 25 to 600 °C. As shown in Fig. 4a, two steps of weight loss were occurred in the TGA figure. A smaller weight loss of ∼13% in the 25–150 °C range was possibly due to the loss of trapped water,38 while the major weight loss (∼50%) occurred in the range 220–340 °C (Fig. 4a) was probably due to a degradation of the polysaccharide structure. Therefore, the weight at 150 °C was defined as 100% dry weight for the next degradation analysis of PTR-EPS1.
 | ||
| Fig. 4 (a) TGA curves of PTR-EPS1 at different heating rates; (b) determination of decomposition characteristic parameters of PTR-EPS1 at a heating rate of 5 °C min−1. | ||
The decomposition characteristics of PTR-EPS1 were analyzed in the 150–600 °C range with different heating rates (Fig. 4b). To avoid the influence of heating rate procedure in determining the three characteristic temperatures including the onset, the maximum and the shift decomposition temperature (T0, Tp, and Ts, respectively), three heating rates were extrapolated to a value where β = 0 for each of the three temperatures (ESI Fig. S2†). T0, Tp, and Ts were determined to be 205.7 °C, 311.4 °C, and 331.2 °C. The corresponding weight loss percentages in these temperatures were represented by WL0, WLp, and WLs (which indicate the weight loss during the temperature transition in the range from 150 to 205.7 °C, from 150 to 311.4 °C, and from 150 to 331.2 °C) were found to be 2.18 ± 0.42, 57.85 ± 1.24 and 79.74 ± 1.02%, respectively. The fastest degradation of PTR-EPS1 was found during a narrow transition temperature of 19.8 °C from Tp 311.4 °C to Ts (331.2 °C). Moreover, a total of 77.56% of this polysaccharide was decomposed within a temperature range of 125.5 °C during the transition from T0 (205.7 °C) to Ts (331.2 °C).
A, and g(α).
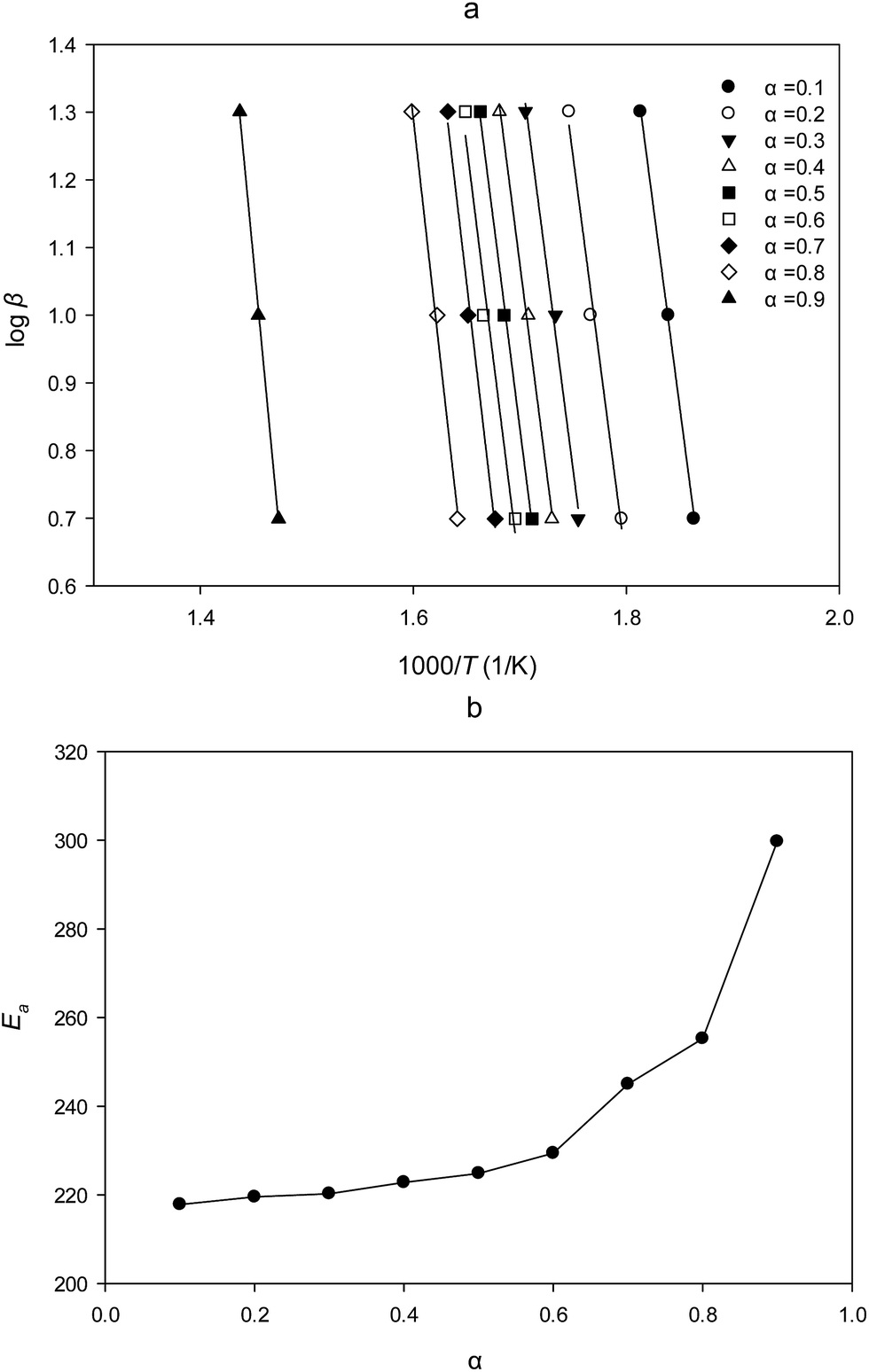 | ||
| Fig. 5 (a) Typical isoconversional plot of FWO method; (b) dependence of Ea on α for PTR-EPS1. | ||
The average apparent activation energy was then calculated to be 223.4 kJ mol−1 from the conversion range of 0.2–0.6 (Table 2). For the fact that any change in lnA shall be accompanied by a corresponding change in Ea, a and b could be obtained by a plot of lnA vs. Ea, derived from Table 3, the experimental lnA was then calculated by use of average FWO activation energy according to eqn (3) as listed in Table 2. The best degradation model was selected on the basis of (i) the correlation coefficient and (ii) the closeness of the activation energy with that determined by FWO method. Therefore, the three-dimensional diffusion model (D3) was found to be the most probable mechanistic function according to the result of Table 2 and the experimental data in Table 3. Then, activation enthalpy (ΔH≠), activation entropy (ΔS≠) and activation free energy (ΔG≠) were obtained according to the thermodynamic eqn (6)–(8) and listed in Table 2.
| Sample | Compensation equation parameters | Ea (kJ mol−1) | lnA |
ΔH≠ (kJ mol−1) | ΔS≠ (J mol−1 K−1) | ΔG≠ (kJ mol−1) | ||
|---|---|---|---|---|---|---|---|---|
| a | b | r | ||||||
| a Note: a and b, compensation parameters of eqn (4); r, the correlation coefficient; Ea, apparent activation energy; A, pre-exponential factor; ΔH≠, activation enthalpy; ΔS≠, activation entropy; ΔG≠, activation free energy. | ||||||||
| PTR-EPS1 | 0.1997 | 10.03 | 0.9984 | 223.4 | 54.64 | 223.4 | 204.2 | 109.9 |
| Reaction model | Codeb | g(α) | Ea | lnA |
r |
|---|---|---|---|---|---|
| a g(α), functional form; α, conversion rate; Ea, A, the calculated apparent activation energy and a pre-exponential factor based on eqn (3); r, the related correlation coefficient.b Code of reaction model. | |||||
| Power law | P1 | α1/4 | 32.7 | 16.16 | 0.9885 |
| Power law | P2 | α1/3 | 40.3 | 17.84 | 0.9267 |
| Power law | P3 | α1/2 | 55.7 | 21.11 | 0.9980 |
| Power law | P4 | α3/2 | 147.6 | 39.98 | 0.9433 |
| One-dimensional diffusion | D1 | α2 | 193.6 | 49.22 | 0.9974 |
| First order | F1 | −ln(1 − α) | 127.1 | 36.35 | 0.9403 |
| Avrami–Erofeyev | A1 | [−ln(1 − α)]1/4 | 39.0 | 17.69 | 0.9583 |
| Avrami–Erofeyev | A2 | [−ln(1 − α)]1/3 | 48.8 | 19.83 | 0.9533 |
| Avrami–Erofeyev | A3 | [−ln(1 − α)]1/2 | 68.4 | 24.04 | 0.9948 |
| Three-dimensional diffusion | D3 | [1 − (1 − α)1/3]2 | 226.5 | 54.27 | 0.9954 |
| Contracting sphere | CS | 1 − (1 − α)1/3 | 118.1 | 33.23 | 0.9657 |
| Contracting cylinder | CC | 1 − (1 − α)1/2 | 115.2 | 32.865 | 0.9647 |
The isoconversional method provides more accurate values of Ea and A than the single heating rate method recommended by International Confederation for Thermal Analysis and Calorimetry (ICTAC).21 The thermal kinetic parameters thus obtained had greater practical significance to understand and predict the thermal degradation process of PTR-EPS1.
Previous studies on thermal decomposition of polymers were mainly focused on natural fibers such as cellulose, hemicellulose, and lignin17 as well as their derivatives (e.g. cyanoethyl cellulose).40 The thermal degradation of hemicellulose and cellulose occurred quickly, with the weight loss of cellulose mainly happened at 315–400 °C and that of hemicellulose at 220–315 °C.41 The activation energy of cellulose and hemicellulose was reported to be 124.42, and 187.06 kJ mol−1, respectively.42 In addition, TGA-DTG was used to determine the thermal stability by monitoring the decomposition process temperature range between nanocomposite and pure drug in a study of 6-mercaptopurine-PEG-coated magnetite nanoparticle delivery system.43 In a recent study, thermal analysis has also been used to determine the long-term stability of a new drug delivery polymer called Quaternary Ammonium palmitoyl Glycol Chitosan (GCPQ) capable of protecting peptides from degradation in the plasma in order to deliver them to the brain.44 Moreover, other thermal degradation parameters including experimental Ea, A and Arrhenius equation, also play an important role in the evaluation of the thermal stability and shelf life of nanoparticles obtained from natural fibers in previous studies of delivery system.17
In TGA figure (Fig. 4a), the major decomposition was considered to be caused by degradation of polysaccharide structure.18 Therefore, the major decomposition temperature range was very important for the prediction of the thermal stability of polysaccharides that used in the design of drug carrier. The temperature range of PTR-EPS1 (205.7–331.2 °C) was found to be consistent with the study of some isolated natural polysaccharides (such as those from Mimosa pudica, Argyreia speciosa, and Acacia modesta), the major weight loss of which occurred in the range 225–325 °C.17 In addition, the present results were consistent with the EPS obtained from other Basidiomycetes mushrooms. For example, EPS from Pseudozyma sp. NII 08165, showed a two-stage thermal degradation with stability up to 220 °C and a degradation temperature of 250 °C.45 EPS produced by Coriolus versicolor was thermally stable up to 280 °C, while the fast weight loss (60%) occurred between 280 °C and 350 °C.46 Cao et al. reported the degradation temperature of the EPS from Pycnoporus sanguineus to be up to 152 °C.47 The differences of thermal degradation behavior among different EPSs might be caused by their different structures and molecular weights. However, previous studies on thermal analysis of mushroom EPS did not give the detailed information of apparent activation energy (Ea) and pre-exponential factor (A), which are the important parameters of Arrhenius equation, where Ea was the minimum energy required to start the decomposition. In the field of drug delivery, it has been reported the mass reduction of drug nanocomposite (FPEGMP-2) starting from 43 °C to 940 °C with four weight loss stages (43–170 °C, 3.7%; 185–334 °C, 6.9%; 329–504 °C, 20.7%; and finally 522–940 °C, 32.8%), which indicated the sustained release of carried drug.43 In a study of synthesized biodegradable nanocomposites, the apparent activation energy was applied to explain the thermal stability of nanoparticle system, which indicated that the degradation starting temperature and degradation activation energies of nanocomposite (PLLA/P-LDH) decreasing as the amounts of drug (P-LDH) increases.48 From the experimental data of thermal analysis, the decomposition model could be deduced by model-free analysis for fitting the functional form [g(α)]. In the present study, the thermal decomposition of PTR-EPS1 fitted the three-dimensional diffusion model with the functional form of g(α) = [1 − (1 − α)1/3]2. In the study of some natural polysaccharides, the obtained functional form was used to predict the lifetime of materials.17 For the potential application in food delivery, the fitted functional form from experimental data may perform an important role in the design of release time. Therefore, the obtained thermal kinetic parameters of PTR-EPS1 have an important reference value for its future application.
Conclusions
In summary, a hyper-branched exopolysaccharide (PTR-EPS1) with spherical structure was firstly obtained by submerged fermentation of mycelium of Pleurotus tuber-regium and characterized as α-mannan consisted of a linear backbone of 1,6-linked α-D-Manp residues with branching points at the O-2 site. The extended spherical conformation of PTR-EPS1 observed by TEM was consistent with its simulated partial 3D structure. Moreover, the intrinsic decomposition parameters (e.g. Ea and A) characterized by isoconversional thermal analysis allow a better understanding of the thermal decomposition behavior of PTR-EPS1 for its pharmaceutical application. As a non-digestible hyper-branched α-mannan, PTR-EPS1 has the potential value for using in food/drug delivery system as natural nanoparticles.Note and references
- J. Van Der Vlist, Polymerization of hyperbranched polysaccharides by combined biocatalysis, University of Groningen, Enschede, The Netherlands, 2011, pp. 87–110 Search PubMed.
- H. Takahashi, S. Sawada and K. Akiyoshi, ACS Nano, 2010, 5, 337–345 CrossRef PubMed.
- Z. H. Liu, Y. P. Jiao, Y. F. Wang, C. R. Zhou and Z. Y. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed.
- B. Hu, Y. Ting, X. Zeng and Q. Huang, Carbohydr. Polym., 2012, 89, 362–370 CrossRef CAS PubMed.
- J. Xiao, S. Nian and Q. Huang, Food Hydrocolloids, 2015, 51, 166–175 CrossRef CAS.
- Y. Kitajyo, Y. Sakai, T. Imai, T. Satoh, H. Kaga and T. Kakuchi, Abstracts of Papers of the American Chemical Society, 2004, 227, U372–U373 Search PubMed.
- L. Q. Yang, S. S. Fu, X. E. Zhu, L. M. Zhang, Y. R. Yang, X. M. Yang and H. Liu, Biomacromolecules, 2010, 11, 3395–3405 CrossRef CAS PubMed.
- Y. Z. Tao, L. N. Zhang and P. C. K. Cheung, Carbohydr. Res., 2006, 341, 2261–2269 CrossRef CAS PubMed.
- S. D. Kulkarni, B. N. Sinha and K. J. Kumar, Int. J. Biol. Macromol., 2015, 72, 1005–1012 CrossRef CAS PubMed.
- W. Q. Huo, W. X. Zhang, W. Wang and X. H. Zhou, Int. J. Biol. Macromol., 2014, 70, 257–265 CrossRef CAS PubMed.
- S. Daus and T. Heinze, Macromol. Biosci., 2010, 10, 211–220 CrossRef CAS PubMed.
- Y. Z. Tao, L. N. Zhang, F. Yan and X. J. Wu, Biomacromolecules, 2007, 8, 2321–2328 CrossRef CAS PubMed.
- L. Chen, B. B. Zhang, J. L. Chen and P. C. K. Cheung, Food Hydrocolloids, 2014, 38, 48–55 CrossRef CAS.
- B. B. Zhang and P. C. K. Cheung, J. Agric. Food Chem., 2011, 59, 1210–1216 CrossRef CAS PubMed.
- B. B. Zhang, L. Chen and P. C. K. Cheung, Biotechnol. Lett., 2012, 34, 1863–1867 CrossRef PubMed.
- B. B. Zhang and P. C. K. Cheung, Bioresour. Technol., 2011, 102, 8323–8326 CrossRef CAS PubMed.
- M. S. Iqbal, S. Massey, J. Akbar, C. M. Ashraf and R. Masih, Food Chem., 2013, 140, 178–182 CrossRef CAS PubMed.
- F. Yao, Q. L. Wu, Y. Lei, W. H. Guo and Y. J. Xu, Polym. Degrad. Stab., 2008, 93, 90–98 CrossRef CAS.
- D. Giron, J. Therm. Anal. Calorim., 2002, 68, 335–357 CrossRef CAS.
- F. Giordano, C. Novak and J. R. Moyano, Thermochim. Acta, 2001, 380, 123–151 CrossRef CAS.
- S. Vyazovkin, A. K. Burnham, J. M. Criado, L. A. Pérez-Maqueda, C. Popescu and N. Sbirrazzuoli, Thermochim. Acta, 2011, 520, 1–19 CrossRef CAS.
- S. Vyazovkin, J. Therm. Anal. Calorim., 2006, 83, 45–51 CrossRef CAS.
- H. D. Hill and J. G. Straka, Anal. Chem., 1988, 170, 203–208 CAS.
- M. DuBois, K. A. Gilles, J. K. Hamilton, P. A. Rebers and F. Smith, Anal. Chem., 1956, 28, 350–356 CrossRef CAS.
- N. Blumenkrantz and G. Asboe-Hansen, Anal. Biochem., 1973, 54, 484–489 CrossRef CAS PubMed.
- L. Chen, W. W. Xu, S. L. Lin and P. C. K. Cheung, Food Hydrocolloids, 2014, 36, 189–195 CrossRef CAS.
- D. Oxley, G. Currie and A. Bacic, Cold Spring Harbor Protocols, 2006, 2006, t4249 CrossRef PubMed.
- CCRC, 2012.
- L. Chen and P. C. K. Cheung, J. Agric. Food Chem., 2014, 62, 2891–2899 CrossRef CAS PubMed.
- J. H. Flynn and L. A. Wall, J. Res. Natl. Bur. Stand., Sect. A, 1966, 70, 487–523 CrossRef CAS.
- T. Ozawa, Bull. Chem. Soc. Jpn., 1965, 38, 1881–1886 CrossRef CAS.
- E. Vinogradov, B. O. Petersen and J. Ø. Duus, Carbohydr. Res., 2000, 325, 216–221 CrossRef CAS PubMed.
- C. Meier, Y. Wu, G. Pramanik and T. Weil, Biomacromolecules, 2014, 15, 219–227 CrossRef CAS PubMed.
- N. Pippa, A. Dokoumetzidis, C. Demetzos and P. Macheras, Int. J. Pharm., 2013, 456, 340–352 CrossRef CAS PubMed.
- C. Demetzos and N. Pippa, Int. J. Pharm., 2014, 473, 213–218 CrossRef CAS PubMed.
- Q. Ding, L. N. Zhang and C. Wu, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 3201–3207 CrossRef CAS.
- A. Parikh and D. Madamwar, Bioresour. Technol., 2006, 97, 1822–1827 CrossRef CAS PubMed.
- M. C. Popescu, C. M. Popescu, G. Lisa and Y. Sakata, J. Mol. Struct., 2011, 988, 65–72 CrossRef CAS.
- G. Varhegyi, M. J. Antal Jr, E. Jakab and P. Szabó, J. Anal. Appl. Pyrolysis, 1997, 42, 73–87 CrossRef CAS.
- A. M. A. Nada and M. L. Hassan, Polym. Degrad. Stab., 2000, 67, 111–115 CrossRef CAS.
- H. Yang, R. Yan, H. Chen, D. H. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- W. Chen and P. Kuo, Energy, 2011, 36, 6451–6460 CrossRef CAS.
- D. Dorniani, A. U. Kura, S. H. Hussein-Al-Ali, M. Z. B. Hussein, S. Fakurazi, A. H. Shaari and Z. Ahmad, Sci. World J., 2014, 2014, 1–11 Search PubMed.
- K. W. Chooi, M. I. Simão Carlos, R. Soundararajan, S. Gaisford, N. Arifin, A. G. Schätzlein and I. F. Uchegbu, J. Pharm. Sci., 2014, 103, 2296–2306 CrossRef CAS PubMed.
- K. V. Sajna, R. K. Sukumaran, L. D. Gottumukkala, H. Jayamurthy, K. S. Dhar and A. Pandey, Int. J. Biol. Macromol., 2013, 59, 84–89 CrossRef CAS PubMed.
- A. P. A. Tavares, M. S. M. Agapito, M. A. M. Coelho, J. A. Lopes Da Silva, A. Barros-Timmons, J. A. J. Coutinho and A. M. R. B. Xavier, World J. Microbiol. Biotechnol., 2005, 21, 1499–1507 CrossRef CAS.
- J. Cao, H. Zhang and C. Xu, J. Taiwan Inst. Chem. Eng., 2014, 45, 2075–2080 CrossRef CAS.
- M. F. Chiang and T. M. Wu, Compos. Sci. Technol., 2010, 70, 110–115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23383j |
| This journal is © The Royal Society of Chemistry 2016 |