
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the agent role of Mn2+ in redirecting the synthesis of Zn(OH)2 towards nano-ZnO with variable morphology†
Daniela
Ghica
 *,
Ioana D.
Vlaicu
*,
Mariana
Stefan
,
Leona C.
Nistor
and
Sergiu V.
Nistor
*,
Ioana D.
Vlaicu
*,
Mariana
Stefan
,
Leona C.
Nistor
and
Sergiu V.
Nistor
National Institute of Materials Physics, Atomistilor Str. 405A, Magurele, 077125, Romania. Web: http://www.lab50.infim.ro Web: http://www.cetresav.infim.roE-mail: ghica@infim.ro; ioana.vlaicu@infim.ro; Fax: +40 21 3690177; Tel: +40 21 2418108 Tel: +40 21 2418290
First published on 3rd November 2016
Abstract
One of the simplest routes to prepare polycrystalline Zn(OH)2 is by coprecipitation, with zinc nitrate as a cation source. However, the addition of even minute amounts of manganese nitrate to the precursors used to prepare pure Zn(OH)2 results in Mn2+ doped nanostructured ZnO. The comparison with other Mn2+ doped metal hydroxides prepared by the same coprecipitation method, involving metal nitrates precursors, shows that this behavior is unique, pertaining only to Zn(OH)2. A systematic study of the samples prepared without and with variable amounts of Mn2+ ions, in the 1 to 5000 ppm nominal concentrations range showed that the re-routing of the reaction takes place even for the lowest nominal dopant concentration of 1 ppm. According to X-ray diffraction, transmission electron microscopy and Fourier transform infrared spectroscopy investigations, both crystallite size and morphology of the resulting nanostructured ZnO samples varied with the Mn2+ nominal concentration. Moreover, quantitative electron paramagnetic resonance investigations showed that the incorporation rate of the Mn2+ ions at different sites in the nanostructured ZnO depended on the nominal Mn2+ concentration. The results are discussed in terms of the coordination properties of the Mn2+ and Zn2+ ions and the nature of the reaction precursors.
1. Introduction
Doped nanocrystalline zinc oxide (nano-ZnO) is one of the most studied semiconductors, many investigations being focused on tailoring the material properties for applications in spintronics, nano- and/or optoelectronics, by changing/adjusting the nature and concentration of the dopant ions.1–4 Doping nanocrystals, typically grown using colloidal synthesis, is still an experimental challenge and the progress in controlling and understanding the incorporation of impurities in semiconductor nanocrystals has been slow.5–7 Thermally induced decomposition of doped precursors has proved to be an alternative efficient way to obtain doped nano-ZnO. Thus, as shown in the case of the Mn2+ doped hydrozincite (ZCB) – Zn5(CO3)2(OH)6 precursor, with proper adjustment of the annealing temperature and atmosphere conditions it was possible to control the size and size distribution of the resulting Mn2+ doped ZnO (ZnO:Mn) nanoparticles.8,9Zinc hydroxide – Zn(OH)2 has attracted a lot of interest during the last years as a precursor in the synthesis of nano-ZnO with controlled size and morphology.10–14 Moreover, as in the case of the ZCB,8,15 doped Zn(OH)2 was expected to be also a valuable precursor for doped nano-ZnO. However, our attempts to prepare Zn(OH)2 by coprecipitation in the presence of Mn2+ ions failed to produce the expected Mn2+ doped Zn(OH)2.13 Every sample prepared in these conditions resulted in nano-ZnO:Mn, independent of the Mn2+ ions concentration. To our knowledge, this is the first observation of a redirection of the Zn(OH)2 synthesis towards nano-ZnO by doping instead of by modifying essential synthesis or post-synthesis parameters (i.e. temperature, pressure, stirring), as previously reported (e.g.ref. 10–14 and 16).
In an effort to understand the extent of the manganese effect on the reaction products and to determine the Mn2+ concentration threshold for the synthesis redirection to take place, we performed a systematic study of the samples prepared without and with a Mn2+ source, varying the nominal Mn2+ ions concentration from 1 to 5000 ppm. Structural and morphological studies have been performed by X-ray diffraction (XRD), transmission electron microscopy (TEM) and Fourier transform infrared (FTIR) spectroscopy. The localization and distribution of the Mn2+ ions in the Zn(OH)2 and ZnO samples were determined by multifrequency electron paramagnetic resonance (EPR) spectroscopy, which we successfully employed to investigate various Mn2+ doped semiconducting nanostructures.8,9,13,17–19 We have shown that EPR spectroscopy is a sensitive and reliable, statistically relevant technique (as it measure the whole volume of the sample), which can offer accurate information concerning the localization of the paramagnetic Mn2+ ions in the volume and at the surface/interface of the nanostructures, the configuration of their neighboring ligands and the local structural/bonding modifications induced by thermo-chemical treatments.
The discussion of the results takes into consideration the role played by the Mn2+ and Zn2+ ions, their favored coordination and the nature of the reaction precursors in redirecting the Zn(OH)2 synthesis towards nano-ZnO.
2. Experimental section
2.1. Synthesis
The starting materials used in the synthesis were analytical grade zinc nitrate hexahydrate (Zn(NO3)2·6H2O – 99% purity), manganese nitrate tetrahydrate (Mn(NO3)2·4H2O – 99.98% purity) and sodium hydroxide (NaOH – 97% purity) from Alfa Aesar, without further purification. Pure crystalline Zn(OH)2 was prepared by coprecipitation of an aqueous solution of zinc nitrate with sodium hydroxide (in excess) by a procedure described elsewhere.13 The syntheses in the presence of Mn2+ ions were made by a similar procedure, which consisted in mixing the corresponding amounts of 0.19 M Zn(NO3)2 and 0.02 M Mn(NO3)2 aqueous solutions at 50 °C to obtain Mn/Zn ratios of 1, 5, 50, 100, 500, 1000 and 5000 ppm (0.5 at%). Afterwards, an aqueous solution of 2.4 M NaOH was added dropwise using a peristaltic pump under continuous stirring. The resulting precipitates were then centrifuged, washed several times with bi-distilled water and finally with absolute ethanol and dried at 60 °C (in air) in a similar way as the pure Zn(OH)2.In order to ascertain the role of the precursor materials in the reactions leading to the final product, several other compounds were synthesized by the same procedure. Thus, to investigate the role of the Zn2+ ions in obtaining ZnO:Mn instead of Mn2+ doped Zn(OH)2, other metal hydroxides such as Mg(OH)2, Ca(OH)2 and Cd(OH)2 were synthesized in both pure and manganese doped forms, with Mn2+ nominal concentrations of 1000 ppm. Also, taking into account the possibility that the anionic part of the precursors (i.e. the nitrate) may be responsible for the observed turn of the reactions, several syntheses were performed starting from M(II) chlorides/acetates (where M(II) = Zn2+ and Mn2+).
2.2. Characterization methods
XRD investigations were performed with a Bruker D8 Advance X-ray diffractometer, in the θ–θ geometry, with Cu anode and Ni filter (λ = 1.54184 Å). The lattice parameters of the identified crystalline phases and the average crystallites size were determined by Rietveld refinement of the experimental data using the Topas v. 3 software from Bruker.TEM/HRTEM morphology and structure investigations at low and high magnifications were performed with an atomic resolution analytical JEOL ARM 200F electron microscope operated at 200 kV, on specimens prepared by crushing the as grown powder in ethanol, dispersing it by sonication, dropping it on lacey carbon grids and drying it at room temperature (RT).
Weighted amounts of pure and Mn doped powdery samples were inserted in calibrated pure fused silica EPR sample tubes of 2 mm and 3 mm inner diameter. The X (9.5 GHz)- and Q (34 GHz)-band EPR measurements were performed at RT with the Bruker ELEXSYS-E580X and -E500Q spectrometers, respectively, from the Center for advanced ESR/EPR techniques (CetRESav – http://www.cetresav.infim.ro/). The X-band spectrometer, equipped with the calibrated Super High QE cylindrical cavity resonator (ER 4123SHQE), was also employed for quantitative determinations of the Mn2+ ions concentration in the investigated samples. The absolute spin quantitation routine (based on the double integration of the spectra), included in the XEPR software from Bruker, was used. The spin Hamiltonian (SH) parameters of the observed paramagnetic centers were determined using a two-steps procedure20 based on the lineshape simulation of the EPR spectra with the EASYSPIN v.5.0.12 software.21
FTIR spectra were recorded on several selected samples with a Spectrum BX II (Perkin Elmer) spectrometer in the 4000–350 cm−1 spectral range, with 128 scans and 4 cm−1 resolution. The ZnO:Mn samples were embedded/diluted in KBr pellets.
3. Results and discussion
3.1. XRD results
The XRD pattern of the undoped sample (Fig. 1 – bottom) shows the presence of a single phase ε-Zn(OH)2 with orthorhombic (P212121) structure [JCPDS card no. 38-0385]. The resulting lattice parameters (Table S1 from ESI†) are similar with those previously reported.13 | ||
| Fig. 1 XRD diffractograms of the undoped (bottom) and Mn2+ doped (upper) samples. | ||
In the case of the Mn2+ doped samples, the analysis of the XRD patterns evidences the formation of a single phase of nanocrystalline hexagonal ZnO [JCPDS card no. 89-1397] in all samples, even for the lowest Mn2+ nominal concentration of 1 ppm. Fig. 1 presents only the XRD patterns of the samples with the minimum and maximum Mn2+ nominal concentration. The resulting ZnO crystallite sizes and lattice parameters for all investigated samples are presented in Table S1 from ESI.† The values are similar for all samples in the 1–1000 ppm nominal concentration range, with notable changes observed only for the sample with the maximum Mn2+ nominal concentration of 5000 ppm: the average crystallite size decreases from 38 nm to 32 nm.
The substitution of the tetrahedrally coordinated Zn2+ ions (Shannon-Prewitt crystal radius RSP = 0.74 Å (ref. 22)) by the larger Mn2+ ions (RSP = 0.80 Å (ref. 22)) produces an expansion of the ZnO lattice associated to lattice disorder. This is evidenced by the changes observed in the XRD parameters, namely an increase of the lattice parameters and a decrease of the crystallite size. Such results have been also reported for other Mn doped ZnO nanostructures (see ref. 18 and references cited therein). If the Mn concentration is lower than 1%, as in our case, the effect is relatively reduced, as presented in Table S1 – ESI.†
There are several mechanisms proposed to explain the inhibition of the ZnO crystallites growth by the presence of impurities.23–25 The reduced diffusivity in ZnO due to Zn concentration decrease in the doped samples suppresses the grain growth. Also, the presence of impurities produces a retarding force on the grain boundaries, impeding the grain growth. Moreover, in Al doped ZnO the grain growth activation energy was determined to increase in the presence of impurities.24 All these would contribute to a gradual decrease of the grain size with the increase of the impurities concentration.
3.2. TEM investigations
TEM images of the undoped Zn(OH)2 sample (see Fig. S1 from ESI†) revealed the plate-like, rod-like morphology of the crystallites of rather large (100–1200 nm) dimensions. The corresponding electron diffraction patterns were indexed with the ε-Zn(OH)2 phase. On the other hand, when the preparation of Mn2+ doped Zn(OH)2 was attempted by the same procedure, the nature/structure of the obtained precipitate changed in a rather drastic way. As shown by X-ray (Fig. 1) and electron diffractions (Fig. 2), even at the smallest (1 ppm) Mn2+ nominal concentration, the ZnO phase was formed instead of Zn(OH)2. | ||
| Fig. 2 TEM/HRTEM images and the corresponding ED patterns (insets) for the ZnO samples doped with 1 ppm (a), 1000 ppm (b), 5000 ppm (c) Mn2+ nominal concentration. (d) HRTEM image at larger magnification of the sample with Mn2+ 5000 ppm nominal concentration. The ED patterns were indexed with the ZnO structure. | ||
The TEM images in Fig. 2 illustrate the changes in the morphology of the resulting ZnO samples doped with Mn2+ ions of increasing nominal concentrations. According to Fig. 2a the sample prepared with 1 ppm Mn2+ nominal concentration consists of large nanoparticles (30–200 nm), most of them with lamellar and rod shapes. It is interesting to note that the pure Zn(OH)2 sample exhibits the same morphology, but the crystallites are much larger (one order of magnitude on the average). In the case of the sample prepared with 1000 ppm Mn2+ nominal concentration (Fig. 2b) the lamellar nanoparticles are smaller, while the rods remain of the same size as in the sample prepared with 1 ppm Mn2+.
Finally, in the case of the sample prepared with 5000 ppm Mn2+ nominal concentration, Fig. 2c shows that the majority of the nanoparticles exhibits a rather uniform morphology with smaller, 20–60 nm size, although some nanorods and lamellar nanoparticles are still present. Therefore, the increase in the Mn2+ nominal concentration leads to the formation of ZnO nanoparticles with much more uniform morphology and smaller dimensions. All the corresponding electron diffraction (ED) patterns in the insets of Fig. 2a–c were indexed with the ZnO structure. The HRTEM image (Fig. 2d) reveals additional details of the sample prepared with 5000 ppm nominal concentration of Mn2+ ions. The morphology of the nanocrystals is indeed polyhedral. Voids with dimensions of 1–16 nm are observed on each nanocrystallite. It looks like a degassing effect took place during their synthesis. Note that the presence of these voids can be also observed in Fig. 2a, in regions where the large crystallites are not overlapped, suggesting that their presence is not related to an increase in the nominal concentration of Mn2+ ions in the solution, but to the synthesis process itself. These voids were observed only in the Mn2+ doped nano-ZnO, independent of the Mn concentration, and not in the undoped Zn(OH)2, as one can see in Fig. S1 from ESI.†
3.3. EPR results
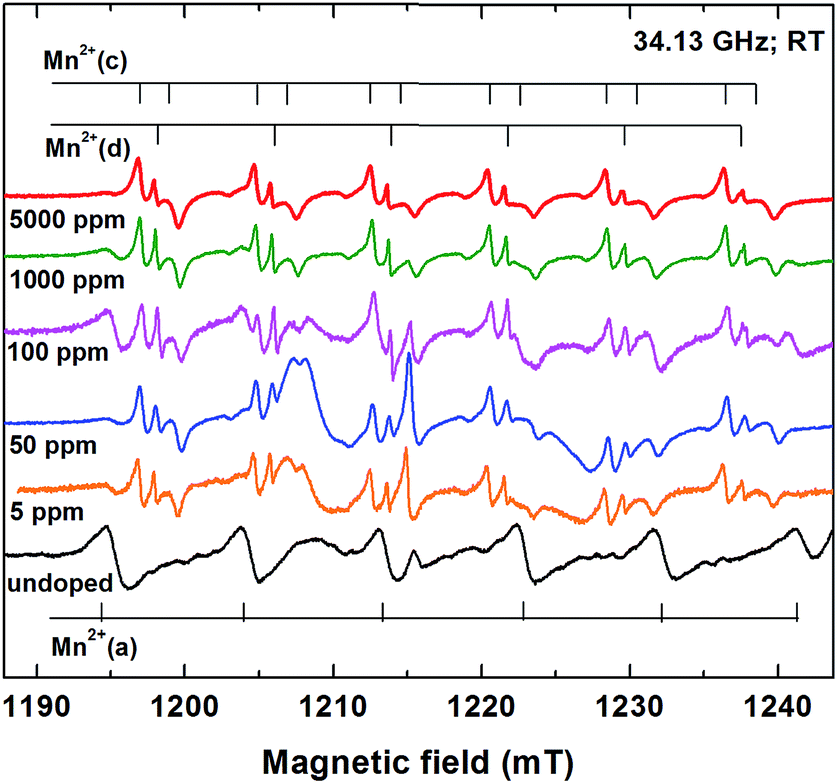 | ||
| Fig. 3 Multiscan Q-band EPR spectra for the undoped (bottom) and Mn2+ doped (upper) samples. The six hyperfine lines/doublet-lines of the Mn2+(a), Mn2+(d)/Mn2+(c) paramagnetic centers are marked with vertical bars. | ||
The EPR spectrum of the undoped sample (Fig. 3 – bottom), recorded with 100 scans, corresponds to paramagnetic impurity levels of less than 1 ppm. It consists of a dominant set of six lines with almost equal intensity, separation (∼9.2 mT) and linewidth, characteristic for the central hyperfine allowed (Ms: −1/2 ↔ 1/2, ΔMI = 0) transitions of the Mn2+ ions (S = 5/2, I = 5/2) in a polycrystalline lattice host.20 A few additional lines from other paramagnetic defects are also present in the EPR spectrum, as previously reported.13
The EPR spectra of the Mn2+ doped samples differ from the EPR spectrum of the undoped sample, due to the different host lattice. These spectra are dominated by six similar groups of three close lying lines, with a ∼7.8 mT separation between the groups, typical for Mn2+ in ZnO.26 Another sextet of lines, with a larger ∼9 mT separation, belonging to a different Mn2+ center, can be observed. The relative intensities of the two sextets are different in the five different Mn2+ doped samples (Fig. 3).
The SH parameters of the Mn2+ centers, determined by simulation and lineshape fitting of the X- and Q-band spectra of the undoped and doped samples, are given in Table S2 from ESI.† In the undoped sample the SH parameters of the paramagnetic center called Mn2+(a) are identical with those previously reported for the substitutional Mn2+ ions in Zn(OH)2.13 The localization of the Mn2+ in the ε-Zn(OH)2 lattice at tetrahedrally, four-fold coordinated Zn2+ cation sites resulted from the analysis of the multifrequency EPR spectra, recorded on as grown and annealed polycrystalline ε-Zn(OH)2.13 One could thus explain why during the thermal decomposition of the polycrystalline ε-Zn(OH)2 into nano-ZnO, the observed paramagnetic Mn2+ centers transformed into Mn2+ centers consisting of Mn2+ ions localized substitutionally at Zn2+ sites in the ZnO lattice.13 The substitutional localization of the Mn2+ impurity ions in ε-Zn(OH)2 at Zn2+ cation sites fourfold coordinated by hydroxyl ligands was further confirmed by the observed changes in the Mn2+ ions hyperfine splitting during the three-steps thermal decomposition of the ε-Zn(OH)2 shell of cubic ZnS nanocrystals with core–shell structure into a ZnO shell.19 The hyperfine splitting changes reflected the sequential dehydration of the four nearest neighbor hydroxyl ligands.
The complex sextet in the EPR spectrum of the Mn2+ doped samples is generated by two types of Mn2+ paramagnetic centers, called Mn2+(c) and Mn2+(d) (see Fig. 3 and 4). Their SH parameters (Table S2 – ESI†) are very close to those previously reported for the Mn2+-c and Mn2+-d centers in the nanostructured ZnO resulted from the thermal decomposition of ZCB,8 which consist of Mn2+ ions substitutionally localized at tetrahedrally coordinated Zn2+ sites in ZnO nanocrystals and disordered ZnO phase, respectively. The very small differences (less than 2%) in the hyperfine parameters of the Mn2+ centers localized in ZnO (see Table S2 – ESI†) can be explained by slightly different average coordination numbers of the Mn2+ ions,9,27 associated with small variations in samples stoichiometry. Such variations could appear due to the particularities of the synthesis routes.
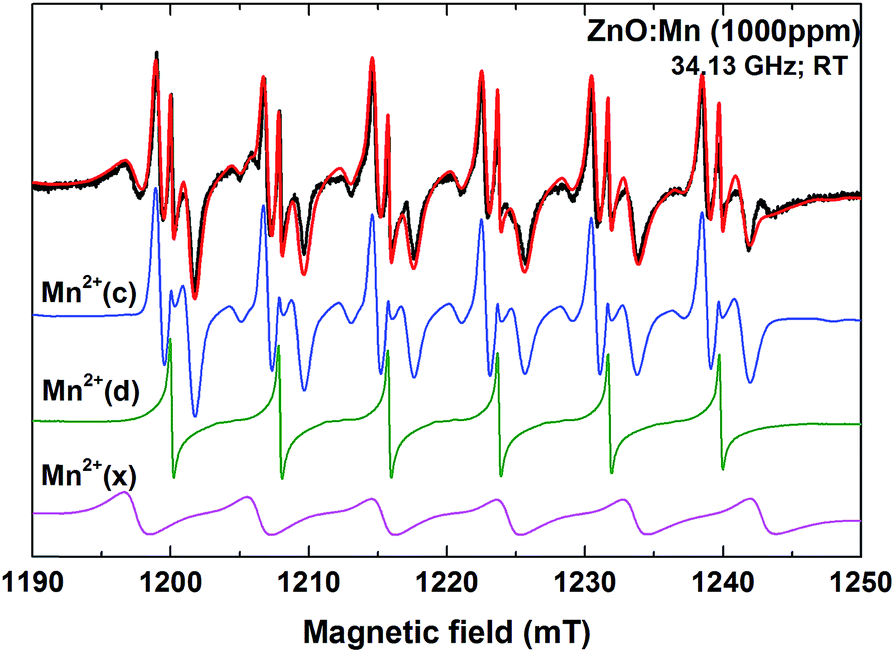 | ||
| Fig. 4 Experimental (black) and simulated (red) Q-band EPR spectrum of the ZnO sample with 1000 ppm Mn2+ nominal concentration. Contributions to the simulated spectrum of the Mn2+ ions localized in crystalline (Mn2+(c) – blue) and disordered (Mn2+(d) – green) ZnO environment, and in the unknown disordered phase (Mn2+(x) – magenta) are shown below. | ||
The third Mn2+ center in the EPR spectrum of the Mn2+ doped samples, with larger hyperfine separation, called Mn2+(x) (Fig. 4), is better observed in the EPR spectrum of the sample with 100 ppm Mn nominal concentration (see Fig. 3). The SH parameters of the Mn2+(x) center, presented in Table S2 from ESI,† indicate the localization of the Mn2+ ions in a disordered environment, different from ZnO.
The evolution of the concentrations of the three Mn2+ centers with the nominal concentration increase, as determined from the deconvoluted spectra, is presented in Table 1.
| Mn2+ nominal concentration | C t [ppm] | Mn2+(c) concentration | Mn2+(d) concentration | Mn2+(x) concentration | |||
|---|---|---|---|---|---|---|---|
| [ppm] | [% Ct] | [ppm] | [% Ct] | [ppm] | [% Ct] | ||
| a The error was estimated to be 25–30%. | |||||||
| 100 ppm | 4 | 1 | 25 | 0.5 | 13 | 2.5 | 62 |
| 500 ppm | 18 | 5 | 29 | 4 | 22 | 9 | 49 |
| 1000 ppm | 26 | 11 | 44 | 5 | 19 | 10 | 37 |
| 5000 ppm | 49 | 25 | 50 | 8 | 16 | 16 | 34 |
Unsurprisingly, the total concentration of incorporated isolated Mn2+ ions is much lower than the nominal concentration, in agreement with the low doping efficiency observed in Mn doped II–VI nanostructures.6,7,28–31 For the samples with nominal concentrations lower than 100 ppm the low incorporation efficiency resulted in very low intensity X-band spectra, and, therefore, in unreliable quantitation results.
One can notice that the percentage of Mn2+(c) centers from the total concentration Ct of isolated Mn2+ ions in the sample increases as the Mn2+ nominal concentration increases. This behavior seems to be correlated with the morphology changes observed by TEM vs. Mn2+ nominal concentration increase, in agreement with theoretical studies which show that the doping efficiency varies with the morphology and size of nanocrystals.5,32
Assuming a uniform distribution of the Mn2+ ions in the sample material, the relative concentrations of the three paramagnetic centers would reflect the proportions of their host structural phases in the sample volume. According to the data in Table 1, the concentration of the Mn2+(x) centers is quite high in all samples, which would correspond to the presence of a large amount of the unknown disordered X phase in the samples. However, no trace of such a secondary phase has been detected by XRD (Fig. 1) or TEM (Fig. 2), meaning that the amount of the disordered X phase is below the detection limit (1–2%). Therefore one concludes that the Mn2+ ions distribution is not uniform in the sample and that the considerably large percentage of Mn2+ ions localized in the disordered phases reflects a segregation process of the Mn2+ ions outside the ZnO nanocrystals. This segregation process diminishes as the nominal concentration of Mn2+ ions increases.
3.4. FTIR measurements
The FTIR spectra recorded for the manganese doped ZnO samples (with 1, 50, 1000, and 5000 ppm Mn nominal concentrations) exhibit similar features, with small differences related to the intensities and positions of certain absorption bands (Fig. 5). The sharp and very intense absorption bands in the 560–400 cm−1 spectral range correspond to the presence of a dominant ZnO phase. The positions and number of these absorption bands, characteristic to the ν(Zn–O) stretching vibration modes, depend on the particles morphology.33,34 Thus, spherical ZnO particles usually show a single distinct IR absorption band, while for ZnO particles with different morphologies this band splits in characteristic ways.33,34 Following the profiles of these IR bands in Fig. 5, one can notice that, by increasing the Mn nominal concentration, the positions, number and intensities of the ν(Zn–O) bands change in agreement with the morphologic changes of the nanostructured ZnO phase, from the dominant lamellar towards the dominant polyhedral shape. These variations in the nanostructured ZnO morphology with the Mn doping concentration, reflected in the FTIR spectra, are in agreement with the TEM results.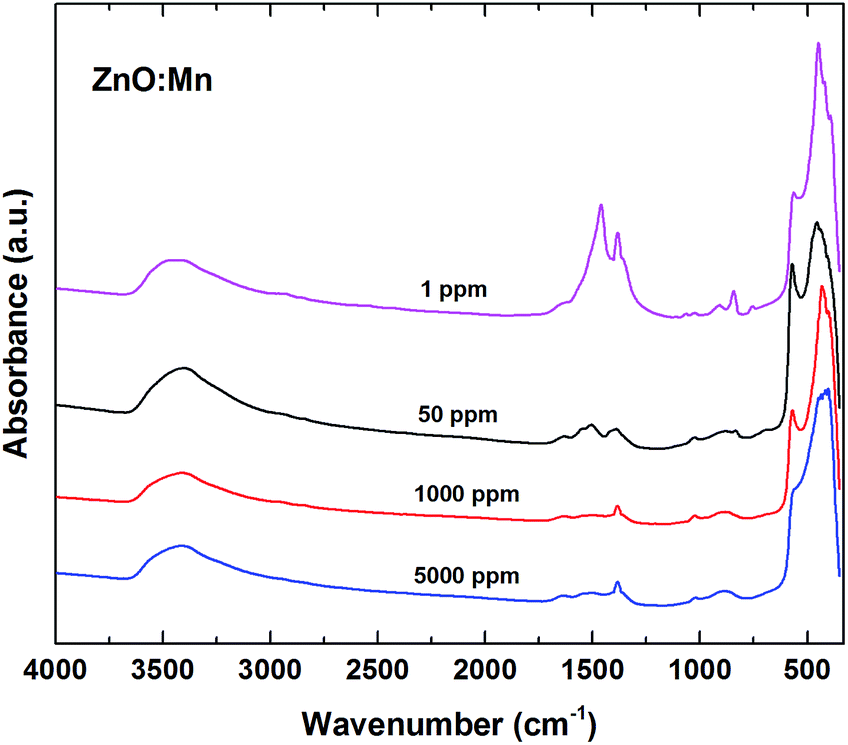 | ||
| Fig. 5 FTIR spectra for ZnO:Mn samples doped with 1, 50, 1000, and 5000 ppm Mn nominal concentration. | ||
Several medium to weak intensity absorption bands observed in the 1460–760 cm−1 spectral range, assigned to the vibration modes for nitrate ions (NO3−), indicate the presence of a secondary phase which contains nitrate groups in its chemical composition.35 The vibration modes observed at ∼3420 cm−1 and ∼1640 cm−1 are attributed to the presence of hydroxyl ions, which can originate from adsorbed water or a hydroxide compound.36 The FTIR results are in agreement with the EPR data, confirming the coexistence in these samples of a dominant ZnO phase (nanocrystalline + disordered) and a secondary X phase (disordered) which, according to FTIR data, is expected to contain nitrate groups. Such a secondary phase could be incorporated between the aggregated ZnO nanoparticles during the final drying step, similar to the case of the cZnS nanoparticles synthesis.37 Table S3 from ESI† summarizes the most important absorption bands maxima observed in the infrared spectra and their assignments for the investigated ZnO:Mn samples.
Although the change in the morphology of the nanostructured ZnO with increasing Mn2+ concentration, observed by TEM and FTIR, is not a new phenomenon, a recently published review38 regarding the influence of ionic impurities on the phase and growth/morphology of semiconductor nanocrystals shows that, despite intensive research, there are fundamental issues still unsolved. Previous articles have reported morphological changes of the ZnO nanomaterials associated with the doping process,39–43 which could be connected to specific structural and coordination properties, as further explained. The crystallographic plane with the minimum surface free energy determines the preferential growth direction. For semiconductors, the surface free energy depends on the hybridized orbit. In the tetrahedrally coordinated (sp3 hybridized orbit) ZnO, with wurtzite structure, the (001) plane has the minimum surface free energy, resulting in a preferential growth along the c-axis, i.e. a (001) “self-texture”.44 The impurities, once added, become involved in the nucleation and growth processes, influencing thus the surface free energy and therefore the morphology and size of a given crystallite. For example, doping induced morphology changes were reported in the case of the ZnO nanorods, with the different shapes depending on the nature of the impurities: better-formed nanorods (Li, Na doped ZnO), petal-like particles (Mg doped ZnO), both rods and irregular shaped particles with a tendency to agglomeration (Cu doped ZnO), spherical particles (Pr3+ doped ZnO).39 Moreover, doping with Fe3+ ions was reported to induce important changes in the morphology of nanostructured ZnO, ranging from nanowires to nanorods and then to nanoparticles, only by increasing the dopant concentration.41 Also, in Ni/Fe mono- or co-doped ZnO nanomaterials, the morphology changed from highly crystalline nanorods at low dopant concentration (<5%) towards well dispersed plate-like nanoparticles at higher dopant concentration.42 The explanation offered for these effects was that the replacement of the Zn2+ ions by Fe2+/Ni2+ ions could affect the local charge density, polarity and potential energy of specific crystal planes, thus resulting in changes of the growth orientation with the Fe/Ni dopant concentration increase.42
Yang et al.40 have shown that ZnO faceted nanocrystals changed into nano-tetrapods when doped with a few percent of divalent Mg, Cd, Ni or Mn ions. At high doping concentrations (>30%), the tetrapods changed into nanowires. They experimentally found that the dopants play an important role in the primary growth stage, resulting in initial growth seeds with diverse crystallographic structures, which are critical for the generation of doped nanocrystals with different shapes. Further investigations dedicated to Mn doped nano-ZnO45 explored the growth mechanism of doped ZnO with an interesting shape transition from tetrapods to spherical nanoparticles by increasing the reaction time and temperature, without changing the dopant concentration. However, it was shown38,40,45 that the growth mechanism is not simple and further experiments and theoretical simulations are required for specific growth conditions.
3.5. Comparison with metal hydroxides prepared by the same method
The most interesting and surprising result of this work is the redirection of the Zn(OH)2 synthesis towards nano-ZnO even for nominal concentrations of Mn as low as 1 ppm added in the synthesis. However, the manganese doping was successfully achieved in other hydroxide materials such as Mg(OH)2, Ca(OH)2, β-Ni(OH)2,46–49 without producing structural changes of the host lattice. EPR studies have been performed and the SH parameters of the Mn2+ paramagnetic centers were determined in Mg(OH)2 and Ca(OH)2 single crystals (SC) doped with significant concentrations of Mn2+ ions. In contrast, the SH parameters of the Mn2+ centers in Zn(OH)2 were determined only recently in polycrystalline Zn(OH)2 containing traces of Mn2+ ions (<1 ppm).13 Because the above mentioned results were obtained on doped crystals synthesized by different procedures than the one we used, we decided to prepare several metal hydroxides, pure and doped with Mn, in order to check for a similar redirection of the hydroxide synthesis towards oxide induced by the Mn presence. We used the same preparation procedure and precursors as for Zn(OH)2, replacing the Zn nitrate with nitrates of Mg, Ca and Cd. Fig. 6 presents the XRD patterns of the undoped and Mn (1000 ppm) doped Mg(OH)2, Ca(OH)2 and Cd(OH)2 samples thus obtained. The XRD diffractograms have been indexed with the Mg(OH)2, Ca(OH)2 and Cd(OH)2 structures, according to the JCPDS card no. 86-0441, JCPDS card no. 87-0674 and JCPDS card no. 73-0969, respectively. It is obvious that doping with 1000 ppm Mn did not induce structural changes of the Mg(OH)2, Ca(OH)2 and Cd(OH)2 lattices, unlike the case of Zn(OH)2. | ||
| Fig. 6 XRD diffractograms of the pure and Mn (1000 ppm) doped Mg(OH)2, Ca(OH)2, and Cd(OH)2 samples. | ||
We have also investigated the possible influence of the anion nature in the starting materials, by replacing the M(II) nitrates with M(II) acetates and chlorides, where M(II) = Zn2+ and Mn2+, and leaving the other parameters of the preparation algorithm unaltered. The synthesis performed in the same conditions using zinc acetate as starting material resulted in a hydroxycarbonate compound, while the one using zinc chloride lead to zinc oxide. On the other hand, there are synthesis conditions (temperature, pH) for which ZnO is obtained from all the precursors, i.e. containing nitrate, chloride, sulphate or acetate.50–53
The addition of the Mn source in the synthesis procedure did not affect the result in either case. However, as none of the four synthesis procedures performed (acetate/chloride precursors with/without manganese) resulted in Zn(OH)2, they cannot shed light on the cause of the synthesis redirection observed for nitrate precursors. Therefore this forms a separate subject which can be further studied.
In the case of the other metal hydroxides, we have to take into consideration that zinc itself could be responsible for the observed effect. Indeed, as previously reported,54–56 Zn2+ has a different behavior than the other metal ions regarding its preferred coordination number and ligands type. While Mn(OH)2, Mg(OH)2, Ca(OH)2, Cd(OH)2 and β-Ni(OH)2 exhibit the same CdI2 type crystal structure (hexagonal system), where each cation is surrounded by six anions forming a slightly compressed octahedron, in the case of Zn(OH)2, the Zn2+ ion has a tetrahedral coordination, each cation being surrounded by four anions. This difference in the coordination behavior of Zn2+ compared to the other M(II) ions could lead to a change in the hydroxide host lattice when the Mn2+ ions enter in Zn2+ sites, thus leading to the formation of doped ZnO instead of doped Zn(OH)2. A possible explanation is that by forcing the Mn2+ ions, which prefer an octahedral coordination in almost all compounds, to replace Zn2+ in tetrahedrally coordinated sites in the Zn(OH)2 lattice, a coordinative hindrance appears for manganese. As the specific role played by Mn2+ is restricted only to Zn(OH)2, it is obvious that the observed effect is based on a fine balance between the Zn2+ cation, nitrate anion and Mn2+ dopant, which makes it difficult to give a precise growth mechanism at this stage. The elucidation of the exact reactions which take place during the synthesis necessitates more in-depth/dedicated investigations. Moreover, in spite of extensive studies, it is difficult to propose a definite growth mechanism even for pure ZnO, since there are many factors (such as temperature, pH, water presence) affecting the reactions.38,57
4. Conclusion
Zn(OH)2 is one of the valued precursor materials which by a simple thermal annealing results in nanostructured ZnO with controlled size and size distribution. The attempt to add “controlled doping” to the properties of the resulting nano-ZnO lead us to the observation that the simple synthesis by coprecipitation method used for the envisaged Mn2+ doped Zn(OH)2 was redirected and the final product was nano-ZnO:Mn. To our knowledge this is the first observation of the Zn(OH)2 synthesis redirection towards ZnO where the agent was a dopant ion instead of a synthesis or post-synthesis parameter such as temperature, pressure or stirring. The systematic investigation of the samples prepared without Mn2+ and with Mn2+ ions in a wide nominal concentrations range showed that the synthesis redirection takes place for a Mn2+ nominal concentration as low as 1 ppm.We have also found out that this effect takes place only for Zn(OH)2, while the synthesis of other M(II) hydroxides (M(II) = Ca2+, Mg2+, Cd2+) by the same procedure is not affected by the presence of a Mn source in the starting material. A possible explanation for the synthesis redirection from Zn(OH)2 towards nano-ZnO is based on the different coordination properties of the Mn2+ and Zn2+ ions, which could lead to a coordinative hindrance for the Mn2+ ions.
We have also examined the extent of the manganese influence on the synthesis products. EPR investigations showed a non-uniform distribution of the Mn2+ ions in the resulting nano-ZnO samples. Besides the Mn2+ ions substitutionally localized in tetrahedrally coordinated Zn2+ sites in nanocrystalline and disordered ZnO, a significant amount of Mn2+ ions are segregated in a disordered secondary phase. FTIR investigations showed that this secondary phase could consist of rests of nitrate compounds from the precursors, in an amount below the detection limit of XRD. It was also found out that the percentage of segregated Mn2+ ions from the total concentration of Mn2+ ions incorporated in the samples decreases with the nominal concentration increase, while the percentage of Mn2+ ions localized in ZnO nanocrystals increases.
TEM and FTIR investigations evidenced morphological changes of the ZnO nanostructures associated with the Mn concentration increase. This result points to an interesting possibility to control the morphology and size of the ZnO nanostructures by varying the doping concentration, which is important for designing ZnO nanoparticles with high surface area for specific applications, such as catalysis or gas sensing.
We believe that this subject merits further experimental and theoretical investigations not least because, in the end, it provides a simple, cost and time effective, non-toxic method of preparation for manganese doped ZnO, with controlled morphology and particle size.
Acknowledgements
This work was supported by ANCS, Core Program project number PN16-480102 and by a grant of the Romanian National Authority for Scientific Research and Innovation, CNCS – UEFISCDI, project number PN-II-RU-TE-2014-4-0939.References
- U. Ozgur, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Dogan, V. Avrutain, S.-J. Cho and H. Morkoç, A comprehensive review of ZnO materials and devices, J. Appl. Phys., 2005, 98, 041301 CrossRef.
- L. Schmidt-Mende and J. L. MacManus-Driscoll, ZnO – nanostructures, defects, and devices, Mater. Today, 2007, 10, 40–48 CrossRef CAS.
- E. Fortunato, A. Gonçalves, A. Pimentel, P. Barquinha, G. Gonçalves, L. Pereira, I. Ferreira and R. Martins, Zinc oxide, a multifunctional material: from material to device applications, Appl. Phys. A: Mater. Sci. Process., 2009, 96, 197–205 CrossRef CAS.
- R. L. Z. Hoye, K. P. Musselman and J. L. MacManus-Driscoll, Research update: doping ZnO and TiO2 for solar cells, APL Mater., 2013, 1, 060701 CrossRef , and references cited therein.
- S. C. Erwin, L. Zu, M. I. Haftel, A. L. Efros, T. A. Kennedy and D. J. Norris, Doping semiconductor nanocrystals, Nature, 2005, 436, 91–94 CrossRef CAS PubMed.
- D. J. Norris, A. L. Efros and S. C. Erwin, Doped nanocrystals, Science, 2008, 319, 1776–1779 CrossRef CAS PubMed.
- S. V. Nistor, M. Stefan, L. C. Nistor, D. Ghica, I. D. Vlaicu and A. C. Joita, Doping ultrasmall cubic ZnS nanocrystals with Mn2+ ions over a broad nominal concentration range, J. Phys. Chem. C, 2015, 119, 23781–23789 CAS.
- S. V. Nistor, L. C. Nistor, M. Stefan, D. Ghica, G. Aldica and J. N. Barascu, Crystallization of disordered nanosized ZnO formed by thermal decomposition of nanocrystalline hydrozincite, Cryst. Growth Des., 2011, 11, 5030–5038 CAS.
- M. Stefan, S. V. Nistor and D. Ghica, Correlation of lattice disorder with crystallite size and the growth kinetics of Mn2+ doped ZnO nanocrystals probed by Electron Paramagnetic Resonance, Cryst. Growth Des., 2013, 13, 1350–1359 CAS.
- R. A. McBride, J. M. Kelly and D. E. McCormack, Growth of well-defined ZnO microparticles by hydroxide ion hydrolysis of zinc salts, J. Mater. Chem., 2003, 13, 1196–1201 RSC.
- S. Music, D. Dragcevic and S. Popovic, Influence of synthesis route on the formation of ZnO particles and their morphologies, J. Alloys Compd., 2007, 429, 242–249 CrossRef CAS.
- M. Wang, Y. Zhou, Y. Zhang, S. H. Hahn and E. J. Kim, From Zn(OH)2 to ZnO: a study on the mechanism of phase transformation, CrystEngComm, 2011, 13, 6024–6026 RSC.
- S. V. Nistor, D. Ghica, M. Stefan, I. Vlaicu, J. N. Barascu and C. Bartha, Magnetic defects in crystalline Zn(OH)2 and nanocrystalline ZnO resulting from its thermal decomposition, J. Alloys Compd., 2013, 548, 222–227 CrossRef CAS.
- F. Demoisson, R. Piolet and F. Bernard, Hydrothermal synthesis of ZnO crystals from Zn(OH)2 metastable phases at room to supercritical conditions, Cryst. Growth Des., 2014, 14, 5388–5396 CAS.
- Z. Mickovic, D. T. L. Alexander, A. Sienkiewicz, M. Mionic, L. Forro and A. Magrez, Synthesis of nanosized Mn-doped ZnO by low temperature decomposition of hydrozincite precursors, Cryst. Growth Des., 2010, 10, 4437–4441 CAS.
- A. Top and H. Cetinkaya, Zinc oxide and zinc hydroxide formation via aqueous precipitation: effect of the preparation route and lysozyme addition, Mater. Chem. Phys., 2015, 167, 77–87 CrossRef CAS.
- D. Ghica, M. Stefan, C. Ghica and G. E. Stan, Evaluation of the paramagnetic impurities segregation at grain boundaries in nanostructured ZnO films, ACS Appl. Mater. Interfaces, 2014, 6, 14231–14238 CAS.
- M. Stefan, D. Ghica, S. V. Nistor, A. V. Maraloiu and R. Plugaru, Mn2+ ions distribution in doped sol-gel deposited ZnO films, Appl. Surf. Sci., 2016 DOI:10.1016/j.apsusc.2016.02.167.
- S. V. Nistor, D. Ghica, M. Stefan and L. C. Nistor, Sequential thermal decomposition of the shell of cubic ZnS/Zn(OH)2 core-shell quantum dots observed with Mn2+ probing ions, J. Phys. Chem. C, 2013, 117, 22017–22028 CAS.
- M. Stefan, S. V. Nistor and J. N. Barascu, Accurate determination of the spin Hamiltonian parameters for Mn2+ ions in cubic ZnS nanocrystals by multifrequency EPR spectra analysis, J. Magn. Reson., 2011, 210, 200–209 CrossRef CAS PubMed.
- S. Stoll and A. Schweiger, EasySpin, a comprehensive software package for spectral simulation and analysis in EPR, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- A. Michels, C. E. Krill, H. Erhardt, R. Birringer and D. T. Wu, Modelling the influence of grain-size-dependent solute drag on the kinetics of grain growth in nanocrystalline materials, Acta Mater., 1999, 47, 2143–2152 CrossRef CAS.
- B. Ingham, R. Linklater and T. Kemmitt, Grain growth kinetics of ZnO: Al nanocrystalline powders during calcination from sol-gel, J. Phys. Chem. C, 2011, 115, 21034–21040 CAS.
- R. Karmakar, S. K. Neogi, A. Banerjee and S. Bandyopadhyay, Structural, morphological, optical and magnetic properties of Mn doped ferromagnetic ZnO thin film, Appl. Surf. Sci., 2012, 263, 671–677 CrossRef CAS.
- M. Diaconu, H. Schmidt, A. Poeppl, R. Bottcher, J. Hoentsch, A. Klunker, D. Spemann, H. Hochmuth, M. Lorenz and M. Grundmann, Electron paramagnetic resonance of Zn1−xMnxO thin films and single crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 085214 CrossRef.
- E. Simanek and K. A. Muller, Covalency and hyperfine structure constant A of iron group impurities in crystals, J. Phys. Chem. Solids, 1970, 31, 1027–1040 CrossRef CAS.
- N. Tsujii, H. Kitazawa and G. Kido, Magnetic properties of Mn- and Eu-doped ZnS nanocrystals, J. Appl. Phys., 2003, 93, 6957–6959 CrossRef CAS.
- R. M. Krsmanovic Whiffen, D. J. Jovanovic, Z. Antic, B. Bartova, D. Milivojevic, M. D. Dramicanin and M. G. Brok, Structural, optical and crystal field analyses of undoped and Mn2+ - doped ZnS nanoparticles synthesized via reverse micelle route, J. Lumin., 2014, 146, 133–140 CrossRef CAS.
- S. Bhattacharyya, D. Zitoun and A. Gedanken, Electron paramagnetic resonance spectroscopic investigation of manganese doping in ZnL (L = O, S, Se, Te) nanocrystals, Nanosci. Nanotechnol. Lett., 2011, 3, 541–549 CrossRef CAS.
- S. V. Nistor, M. Stefan, L. C. Nistor, D. Ghica and I. D. Vlaicu, Distribution and interaction of Mn2+ ions incorporated in cubic ZnS quantum dots over a broad concentration range, J. Alloys Compd., 2016, 662, 193–199 CrossRef CAS.
- T. Singh, T. J. Mountziaris and D. Maroudas, On the transition-metal doping efficiency of zinc oxide nanocrystals, Appl. Phys. Lett., 2010, 97, 073120 CrossRef.
- M. Andrés Vergés, A. Mifsud and C. J. Serna, Formation of rod-like zinc oxide microcrystals in homogeneous solutions, J. Chem. Soc., Faraday Trans., 1990, 86, 959–963 RSC.
- F. A. Sigoli, M. R. Davolos and M. Jafelicci, Morphological evolution of zinc oxide originating from zinc hydroxide carbonate, J. Alloys Compd., 1997, 262–263, 292–295 CrossRef CAS.
- J. Coates, Interpretation of infrared spectra, a practical approach, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, Wiley and Sons Ltd., Chichester, 2000, pp. 10815–10837 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 1986 Search PubMed.
- L. C. Nistor, C. D. Mateescu, R. Birjega and S. V. Nistor, Synthesis and characterization of mesoporous ZnS with narrow size distribution of small pores, Appl. Phys. A: Mater. Sci. Process., 2008, 92, 295–301 CrossRef CAS.
- A. K. Guria and N. Pradhan, Doped or not doped: ionic impurities for influencing the phase and growth of semiconductor nanocrystals, Chem. Mater., 2016, 28, 5224–5237 CrossRef CAS.
- K. Jayanthi, S. Chawla, K. N. Sood, M. Chhibara and S. Singh, Dopant induced morphology changes in ZnO nanocrystals, Appl. Surf. Sci., 2009, 255, 5869–5875 CrossRef CAS.
- Y. Yang, Y. Jin, H. He, Q. Wang, Y. Tu, H. Lu and Z. Ye, Dopant-induced shape evolution of colloidal nanocrystals: the case of zinc oxide, J. Am. Chem. Soc., 2010, 132, 13381–13394 CrossRef CAS PubMed.
- J. Iqbal, T. Jan, Y. Ronghai, S. H. Naqvi and I. Ahmad, Doping induced tailoring in the morphology, band-gap and ferromagnetic properties of biocompatible ZnO nanowires, nanorods and nanoparticles, Nano-Micro Lett., 2014, 6, 242–251 CrossRef.
- C. Fletcher, Y. Jiang, C. Sun and R. Amal, Morphological evolution and electronic alteration of ZnO nanomaterials induced by Ni/Fe co-doping, Nanoscale, 2014, 6, 7312–7318 RSC.
- P. M. Shirage, A. K. Rana, Y. Kumar, S. Sen, S. G. Leonardi and G. Neri, Sr- and Ni-doping in ZnO nanorods synthesized by a simple wet chemical method as excellent materials for CO and CO2 gas sensing, RSC Adv., 2016, 6, 82733–82742 RSC.
- N. Fujimura, T. Nishihara, S. Goto, J. Xu and T. Ito, Control of preferred orientation for ZnOx films: control of self-texture, J. Cryst. Growth, 1993, 130, 269–279 CrossRef CAS.
- Y. Yang, Y. Li, L. Zhu, H. He, L. Hu, J. Huang, F. Hu, B. He and Z. Ye, Shape control of colloidal Mn doped ZnO nanocrystals and their visible light photocatalytic properties, Nanoscale, 2013, 5, 10461–10471 RSC.
- W. A. Pieczonka, H. E. Petch and A. B. McLay, An electron spin resonance study of manganese impurity in brucite, Can. J. Phys., 1961, 39, 145–157 CrossRef CAS.
- F. Holuj, S. M. Quick and M. Rosen, Vibronic effects in the electron paramagnetic resonance of Mn2+ in Ca(OH)2, Phys. Rev. B: Condens. Matter Mater. Phys., 1972, 6, 3169–3179 CrossRef CAS.
- L. Guerlou-Demourgues and C. Delmas, Electrochemical behavior of the manganese-substituted nickel hydroxides, J. Electrochem. Soc., 1996, 143, 561–566 CrossRef CAS.
- M. Morishita, S. Ochiai, T. Kakeya, T. Ozaki, Y. Kawabe, M. Watada, S. Tanase and T. Sakai, Structural analysis by synchrotron XRD and XAFS for manganese-substituted α - and β -type nickel hydroxide electrode, J. Electrochem. Soc., 2008, 155, A936–A944 CrossRef CAS.
- R. Wahab, S. G. Ansari, Y. S. Kim, M. Song and H.-S. Shin, The role of pH variation on the growth of zinc oxide nanostructures, Appl. Surf. Sci., 2009, 255, 4891–4896 CrossRef CAS.
- A. Bagabas, A. Alshammari, M. F. A. Aboud and H. Kosslick, Room-temperature synthesis of zinc oxide nanoparticles in different media and their application in cyanide photodegradation, Nanoscale Res. Lett., 2013, 8, 516 CrossRef PubMed.
- A. M. Pourrahimi, D. Liu, L. K. H. Pallon, R. L. Andersson, A. Martinez Abad, J.-M. Lagaron, M. S. Hedenqvist, V. Strom, U. W. Gedde and R. T. Olsson, Water-based synthesis and cleaning methods for high purity ZnO nanoparticles – comparing acetate, chloride, sulphate and nitrate zinc salt precursors, RSC Adv., 2014, 4, 35568–35577 RSC.
- M. F. Khan, A. H. Ansari, M. Hameedullah, E. Ahmad, F. M. Husain, Q. Zia, U. Baig, M. R. Zaheer, M. M. Alam, A. M. Khan, Z. A. AlOthman, I. Ahmad, G. Md Ashraf and G. Aliev, Sol-gel synthesis of thorn-like ZnO nanoparticles endorsing mechanical stirring effect and their antimicrobial activities: Potential role as nano-antibiotics, Sci. Rep., 2016, 6, 27689 CrossRef CAS PubMed.
- J. P. Glusker, A. Kaufman Katz and C. W. Bock, Metal ions in biological systems, Rigaku J., 1999, 16, 8–16 CAS.
- C. W. Bock, A. Kaufman Katz, G. D. Markham and J. P. Glusker, Manganese as a replacement for magnesium and zinc: functional comparison of the divalent ions, J. Am. Chem. Soc., 1999, 121, 7360–7372 CrossRef CAS.
- I. Herber, W.-K. Tang, H.-Y. Wong, T.-W. Lam, C.-K. Siu and M. K. Beyer, Reactivity of hydrated monovalent first row transition metal ions [M(H2O)n]+, M = Cr, Mn, Fe, Co, Ni, Cu, and Zn, n < 50, toward acetonitrile, J. Phys. Chem. A, 2015, 119, 5566–5578 CrossRef CAS PubMed.
- E. A. Meulenkamp, Synthesis and growth of ZnO nanoparticles, J. Phys. Chem., 1998, 102, 5566–5572 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23065b |
| This journal is © The Royal Society of Chemistry 2016 |