
New biobased tetrabutylphosphonium ionic liquids: synthesis, characterization and use as a solvent or co-solvent for mild and greener Pd-catalyzed hydrogenation processes†
Safa Hayounia,
Anthony Roberta,
Nadège Ferlina,
Hassen Amrib and
Sandrine Bouquillon*a
aInstitut de Chimie Moléculaire de Reims, UMR CNRS 7312 – Université de Reims Champagne-Ardenne, Boîte No. 44, B. P. 1039, F-51687 Reims, France. E-mail: sandrine.bouquillon@univ-reims.fr; Fax: +33 0 3-26-91-31-66; Tel: +33 0 3-26-91-89-73
bUniversité de Tunis El Manar, Faculté des Sciences de Tunis, Département de Chimie, Laboratoire de Synthèse Organique Sélective & Activité Biologique, Campus Universitaire, 2092 Tunis, Tunisia
First published on 23rd November 2016
Abstract
Phosphonium-based Ionic Liquids (PhosILs) with natural organic derived anions (L-lactate, L-tartrate, malonate, succinate, L-malate, pyruvate, D-glucuronate, D-galacturonate, ferulate, p-coumarate) were easily prepared by acid–base method from tetrabutylphosphonium hydroxide and an excess of the corresponding acid with good yields. Their characterization was realized through classical NMR, IR and elemental analysis techniques; their viscosity and ATG parameters were also determined. These ionic liquids showed good performance and recyclability in the selective Pd-catalyzed hydrogenation of alkenes, polyenes like linoleic acid and enantioselective hydrogenation of unsaturated ketones such as isophorone at room temperature under atmospheric H2 pressure. Furthermore, NMR studies leading to computational calculations were performed to establish easily the composition of the resulting mixture obtained through the hydrogenation of linoleic acid.
Introduction
Ionic liquids (ILs) present a class of salts, usually characterized by low melting points below 100 °C.1 With their numerous properties, ILs qualify as a good candidate to replace classical organic solvents in many fields:1b electrochemistry, organic synthesis, catalysis, complexation, extraction, etc. Compared to their ammonium analogues, ILs containing phosphonium cations are much rarer and studies involving quaternary phosphonium systems are scarce.2 Indeed, ILs based on quaternary nitrogen cations such as imidazolium and pyridinium have been extensively investigated by comparison to phosphonium ILs which have received scant attention; however, there is no predilection for either family as both have their own advantages and disadvantages.2Typical phosphonium cations have the general formula [R′PR3]+, in which three of the alkyl groups are identical while the fourth is different. As well, asymmetric tertiary phosphines (RR′2P or R2R′P) could lead to [RR′2R′′P]+ and [R2R′R′′P]+ cations through free radical addition.3 These phosphonium salts associated to halides are commercially available. Through anion exchange reactions, a large variety of phosphonium-based ILs have been synthetized.4 Halogen free systems can be also produced by direct reaction of tertiary phosphines with alkylating agents such as benzenesulfonate, alkyltosylates, trialkylphosphates, and dialkylsulfates.5 Chiral,6 protic7 and cyclic8 phosphonium based ILs have also been prepared through classical methods used with ammonium analogues. Respective properties (stability, conductivity, self-diffusion) of both ILs families were also compared and revealed important influence of the nature of the cation.
However, classical ammonium or phosphonium based ILs present toxicity on diverse microbial strains and are low biodegradable,9 limiting their character of “green compounds or green solvents”. Consequently, greener ILs have been then extensively investigated since 2010 essentially concerning ammonium, imidazolium, thiazolium, choline or betaine cations associated to aminoacids or esters as counter ions.10 Concerning phosphonium cations, alkyltrihexylphosphonium acesulfamates were obtained in high yields by a metathesis route, starting with alkyltrihexylphosphonium chlorides or bromides and the commercially available acesulfamate potassium salt considered as a nontoxic organoanion.11 This IL was essentially used in electrochemistry. Ohno and coll. also described the synthesis of ILs with 20 natural aminoacids with ammonium12 and phosphonium13 cations. However, in general, the phosphonium analogues showed relatively low levels of biodegradability, in contrast to dialkylimidazolium and alkylpridinium ILs with incorporated ester moieties and octylsulfate anions, as demonstrated by Scammells et al.14
For our part, we prepared a few years ago, ILs with a bioderivated anion, such as acids obtained from biomass (lactic, tartaric, malic… acids), osidic acids or amino acids like proline or derivatives (Fig. 1).15a,b These ILs derived from TBA (tetrabutylammonium) showed good performance and recyclability in catalytic selective hydrogenation of 1,5-cyclooctadiene (1,5-COD) into cyclooctene at room temperature under atmospheric H2 pressure and were less toxic to E. coli than commercial TBABr. Our system was already largely competitive compared to other selective hydrogenation processes of 1,5-COD conducted under harsh conditions (5–50 bar, 50–100 °C and 4–6 h) with different systems BMIM·PF6/Na2[Ru6C(CO)16],16a Ni catalyst coated with BMIM·C8H17SO4 (ref. 16b) and BMIM·BF4/Pd(acac)2.16c
 | ||
| Fig. 1 Biobased TBA ionic liquids. | ||
In fact, catalytic hydrogenations in ILs have become the subject of extensive studies since 1995 triggered by the simultaneous work of Chauvin17a and Dupont.17b Ever since, catalytic reactions involving metal complexes in ILs have been actively investigated. Around 300 ILs have been screened and have led to the production of useful products and intermediates.18 The majority of these ILs contained heterocyclic cations, such as pyridinium, imidazolium, polyalkylammonium and recently synthesized guanidinium, piperidinium, pyrrolium, pyrrolidinium, morpholinium, cholinium, piperazinium, thiazolium. Hydrogenation processes in ILs have been recently reviewed by Gathergood et al.19 in a quite complete book chapter.
Many organometallic catalytic systems were already described for the hydrogenation of isophorone, essentially based on palladium derivatives associated to chiral ligands. These systems consist on black palladium or palladium supported over Al2O3, MgO, BaCO3, SrCO3, CaCO3 or modified silica, associated to proline,20 proline-based chiral modifiers,20h,21 or (−)-dihydroapovincaminic acid ethyl ester in high pressure conditions.22 Other organometallic systems are used like Ru-23 or RANEY® Ni-based24 catalytic systems which preferentially lead to trimethylcyclohexanol (TMCH), wahile Cr–Ni-,25 Zn-26 or Rh-27 based ones yield only the saturated ketone. In general, these catalytic systems are performed in harsh reaction conditions (very high pressure, toxic solvents).20–22 scCO2 as a reaction medium as a replacement for organic solvents for heterogeneously catalyzed hydrogenation of carvone was also highlighted by enhancing conversion.28
Apart from our work with ILs or CILs (Chiral Ionic Liquids),15a,b to our knowledge only one work concerning hydrogenation process in biobased ILs has been reported.15c,29 It concerned γ-valerolactone-based ILs used for transfer hydrogenation, so without the use of metallic species. These ILs were successfully applied as alternative solvents for homogenous catalytic transfer hydrogenation of acetophenone and its substituted forms, as well as other functionalized ketones and alkenes.
Chiral ionic liquids (CILs) tetrabutylammonium-(S)-prolinate, tetrabutylammonium-(R)-prolinate and tetrabutylammonium trans-4-hydroxy-(S)-prolinate were also easily prepared by our team in one step from the aminoacid and tetrabutylammonium hydroxide (TBAOH) and revealed interesting properties in ecotoxicity and in asymmetric catalysis of α,β-unsaturated ketones such as isophorone.15b
Furthermore, as hydrogenation of vegetable oils represents a real challenge, we were interested to develop these hydrogenation processes in ILs. In 2012, Pagliaro and co-workers prepared some Pt(0) nanoparticles stabilized on silica which could lead in mild conditions (RT, 1 atm H2) to the complete hydrogenation of vegetal oils for many runs.30 Electrochemical pathways were also performed using Pd/Al2O3 and Pd–Co/Al2O3 as catalysts in various pH conditions: oleic acid was obtained with 80% yield at pH equal to 13.31a The partial hydrogenation of this polyunsaturated acid was also performed by Pd/Al2O3 doped with various thiolate self-assembled monolayers under high H2 pressure and at high temperature.31b
So, this paper covers a detailed description of tetrabutylphosphonium based ionic liquids (PhosILs) and their major influence on hydrogenation catalysis especially their contribution to selective mild hydrogenation procedures of fatty acids or to asymmetric catalysis of α,β-unsaturated ketones.
Results and discussion
Synthesis of phosphonium-based ionic liquids (PhosILs) with tetrabutylphosphonium (TBP) as cation
As previously described for ammonium based ILs,15a PhosILs were prepared from tetrabutylphosphonium hydroxide (TBPOH) and an acid following an acid–base method developed by Ohno and co-workers.12 The acids issued from the biomass were L-lactic, L-tartaric, malonic, succinic, L-malic, pyruvic, D-glucuronic, D-galacturonic acids and S-proline (Scheme 1). Three different pathways were improved considering the different physical properties (solubility, melting points, etc.) of the employed acids. All pathways were simple and efficient, as corresponding ILs were obtained with high yields and most of them were liquid at room temperature (Table 1). | ||
| Scheme 1 Synthesis of PhosILs. | ||
| R–COOH | Pathway/yield (%)/aspect (at RT) | PhosILs |
|---|---|---|
| L-Lactic acid | A/87/oil |  |
| L-Tartaric acid | A/81/wax |  |
| L-Malonic acid | A/92/solid |  |
| L-Succinic acid | A/91/viscous oil |  |
| L-Malic acid | A/92/oil |  |
| Pyruvic acid | B/87/oil |  |
| D-Glucuronic acid | C/80/viscous oil |  |
| D-Galacturonic acid | C/77/wax |  |
| S-Proline | A/87/oil |  |
| R-Proline | A/94/oil |  |
| 4-Hydroxy-S-proline | A/90/oil |  |
These PhosILs have been characterized by usual analytical methods involving NMR, IR and elemental analysis. Thermogravimetric and DSC analyses were also performed and allowed respectively the determination of the decomposition and glass transition temperatures.
Decomposition temperatures were relatively high, up to 315 °C and furthermore higher than ammonium analogs15a except for osidic acid derivatives (7 and 8). These sugar derivatives generally present low thermal stability limited by the strength of heteroatom/heteroatom associations and carbon/hydrogen bonds.32a The higher thermal stability of the synthesized PhosILs was expected, as phosphoniums ILs are generally described in the literature as having better thermal stability than imidazolium or ammonium ILs.32b Therefore, PhosILs 1–6 could play an important role as solvent for different chemical reactions.
The glass transition temperature (Tg) values were all under 0 °C. A negative Tg is classical for ionic liquids. But the fact that negative Tgs were observed with osidic carboxylates derivatives 7 and 8 was different from what was previously found with ammonium analogs, which presented Tgs above 0 °C.15a
Finally, the viscosity was following the relationship between the number of carbon and/or hydrogen bonds as described in the literature32c and it was also influenced by the nature of the anions. Values at 80 °C were in general quite low, except for compounds 2 and 8, which values were 268 cP and 617 cP respectively and lower than their TBA analogues (Fig. 2). ILs with low viscosity are expected, to avoid difficulties of mixing homogeneously or dispersing reactants during reaction (Table 2).
 | ||
| Fig. 2 Viscosity (cP) of TBA and TBP ionic liquids at 80 °C. | ||
| PhosILs | Viscosity (cP) at 80 °C | Tg (°C) | Tdec (°C) |
|---|---|---|---|
| 1 | 21 | — | 314 |
| 2 | 268 | −20 | 167 |
| 3 | — | −9.8 | 288 |
| 4 | 125 | −49.3 | 277 |
| 5 | 105 | −40.2 | 203 |
| 6 | 16 | −59.3 | 315 |
| 7 | — | −8.7 | 130 |
| 8 | 617 | −22.5 | 142 |
| 9 | 47 | −56.8 | 301 |
| 10 | — | — | 241 |
| 11 | — | — | 278 |
Other biobased acids were also employed, as p-coumaric or ferulic acids. p-Coumaric can be found in a wide variety of edible plants such as peanuts or vegetables (navy beans, tomatoes and carrots), wine and also barley grain.23 Ferulic acid (FA), an ubiquitous natural phenolic phytochemical is present in seeds, leaves, both in its free form and covalently conjugated to the plant cell wall polysaccharides, glycoproteins, polyamines, lignin and hydroxy fatty acids.34
The experimental pathway had to be modified taking in account the physical properties of the phenolic acids (Scheme 2) and the synthesis was extended to tetrabutylammonium (TBA) salts to complete the preliminary study.15a
 | ||
| Scheme 2 Synthesis of ILs with p-coumarate and ferulate anions. | ||
The respective salts were obtained with good yields and were all solid at room temperature. But it is more adequate to consider them as molten salts due to their high melting point which is higher than 100 °C. Their decomposition temperatures were also relatively low fluctuating between 132 to 181 °C. As a result, they won't be considered for the hydrogenation studies (Table 3).
| ILs | Yield% | Aspect melting point | Tdec (°C) |
|---|---|---|---|
 |
86 | White solid 115 °C | 132 |
 |
89 | White solid 119 °C | 157 |
 |
89 | White solid 145 °C | 156 |
 |
93 | White solid 150 °C | 181 |
Catalytic hydrogenation
 | ||
| Scheme 3 Hydrogenation of 1,5-COD in PhosIL/water. | ||
In the absence of an IL, complete conversion of 1,5-COD into COA was obtained in water in the first run. Recycling the Pd catalyst led to the formation of COE until 90% with conversion attained at the eight run (Fig. 3a).
 | ||
| Fig. 3 Pd-catalyzed hydrogenation of COD in water (a) and in TBABr/H2O (b). | ||
Similar results were obtained in commercial TBPOH, while hydrogenation reactions in TBPBr (Fig. 3b) led also to COE but with lower conversions each run after the other and generation of isomerization compounds. This phenomenon was already observed in presence of other copper or zinc-based catalysts (CuO–Al2O3, CuO–SiO2 or ZnO–Al2O3) via water gas shift reaction.35 These results confirmed the importance of the nature of the counter anion on the hydrogenation mechanism.
Next, in the presence of PhosILs 2–7, the results were close to those obtained with water as solvent. But improvements were observed as no isomerization compounds traces were present, as shown in Fig. 4a with compound 3/water system.
 | ||
| Fig. 4 Pd-catalyzed hydrogenation of COD in TBP malonate 3/H2O (a) and in TBP lactate 1/H2O (b). | ||
On the contrary, with PhosIL 1/H2O, COA was mainly obtained for all cycles (Fig. 4b). This result was probably due to the low viscosity of TBP L-lactate 1 with regard to the other ILs tested. Indeed, even if few data in the literature are available on the solubility of H2 in ILs, the diffusion of gases depends generally on the viscosity of the environment.36
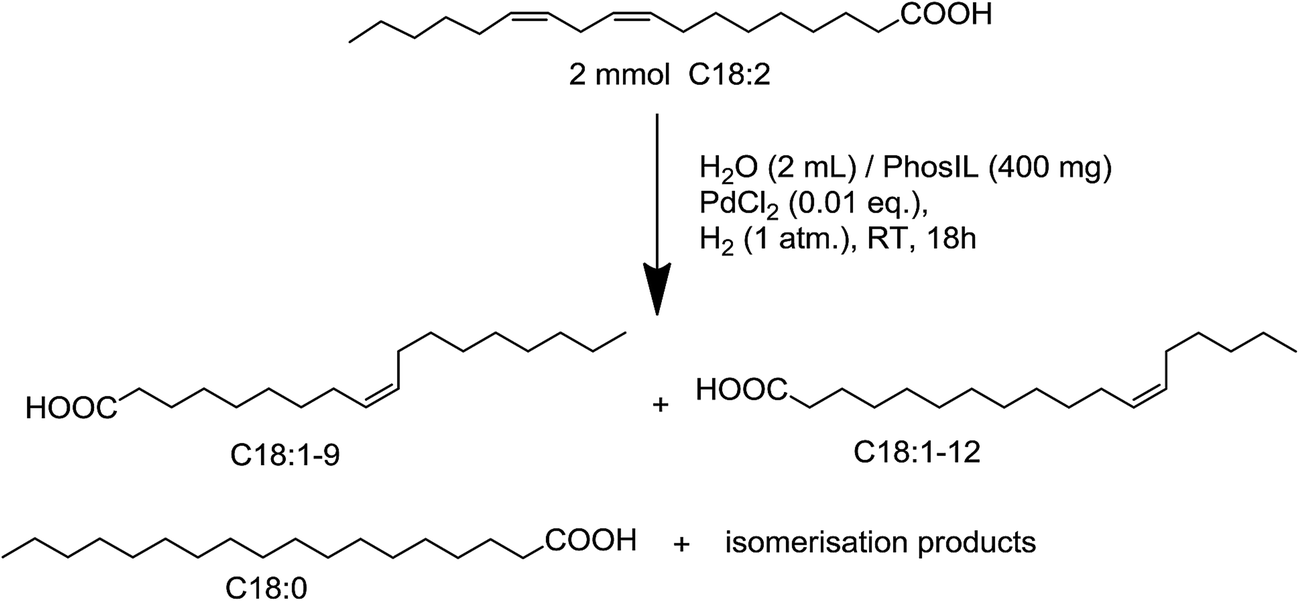 | ||
| Scheme 4 Hydrogenation of linoleic acid in H2O/PhosIL. | ||
The focus of our laboratory was to develop a simple and mild process using the synthesized biosourced PhosILs with water as co-solvent to induce partial hydrogenation and recycling (Scheme 4).
The identification of the composition of the resulting mixture, was assured using gas chromatography (GC), completed by NMR techniques. By GC, after post-hydrogenation esterification of the acids,37 various columns (column TR-1: 100% dimethyl polysiloxane, column TR-5: 5% phenyl methyl polysiloxane, INNOWAX (PEG)) and conditions38 were tested without success to separate and quantify specifically mono and poly-unsaturated isomers presented in the resulting mixture. To solve this problem, a NMR method was developed in our laboratory inspired from reported works concerning the determination of the composition of plant oils.39 Mixtures obtained after hydrogenation were then analyzed by 1H NMR without preliminary separation of constituents.
Assignment of proton signals. The products of linoleic acid hydrogenation in ILs were saturated acid (X), mono-unsaturated acid (Y) and poly-unsaturated acid (Z).
The 1H NMR spectrum of linoleic acid hydrogenation product in ILs is shown in Fig. 5. The spectrum signals are annotated from (a) to (g).39b These groups are assigned as follows: (a), olefenic protons; (b), divinyl methylene protons; (c), α-methylene protons adjacent to carbonyl carbon; (d), allyl methylene protons; (e), β-methylene protons adjacent to carbonyl carbon; (f), methylene protons on saturated carbon atoms; (g), terminal methyl protons of saturated, monounsaturated and polyunsaturated acids; (g′), terminal methyl protons of residual PhosIL traces.
 | ||
| Fig. 5 1H NMR spectrum in CDCl3 of the mixture obtained after hydrogenation of linoleic acid in TBP lactate 1/H2O. | ||
In the divinyl methylene protons chemical shift, there were three signals (Fig. 6). The triplet at 2.77 ppm corresponded to linoleic acid divinyl methylene protons and was confirmed by NMR spectrum of linoleic acid. The other signals were attributed to isomer product of linoleic acid divinyl methylene protons.
 | ||
| Fig. 6 Divinyl methylene 1H NMR spectrum in CDCl3 of hydrogenation product in PhosILs/H2O. (Triplet at 2.77 ppm corresponded to linoleic acid divinyl methylene protons.) | ||
After calculations (see ESI†), composition of hydrogenated products could be determined. The analytical results for the hydrogenation of linoleic acid in TBP malonate 3/H2O can be summarized on the following graph (Fig. 7). Selective hydrogenation towards mono-unsaturated compounds Y was observed with almost complete conversion of the linoleic acid. The selective hydrogenation was also realized without formation of compounds presenting a trans configuration as observed with NMR coupling constants. Moreover, catalytic system was could be easily recycled and reused without changes in reactivity for 6 cycles. Results were strongly similar regardless of the nature of the used PhosILs. Contrary to what was previously observed with 1,5-COD, it seemed there was no effect of neither the anion nor the viscosity of the used PhosILs for hydrogenation of linoleic acid.
 | ||
| Fig. 7 Hydrogenation of linoleic acid in TBP malonate 3/H2O. | ||
The results obtained during our study in soft conditions (RT, atmospheric pressure, water) were quite near to those obtained by Suarez and coll. in 2011 for the selective hydrogenation of biodiesel, however a drastic system (Pd(OAc)2/[BMIM] [BF4], H2 (5–75 bar) and 80 °C) was used.40
As a fair assessment, it is safe to say that our system of hydrogenation was very competitive towards all systems of hydrogenation described until today in the literature for such compounds. It was realized under 1 atm of H2 at room temperature and did not require preliminary preparation of catalysts. The selective hydrogenation of the linoleic acid towards mono-unsaturated compounds was also realized without formation of compounds presenting a trans configuration as observed with NMR coupling constants.
Isophorone was chosen as model substrate to define the best conditions. The conversion and the selectivity were determined by GC; the determination of the enantiomeric excess (ee) requiring the use of a β-cyclodextrine chiral column.
First of all, the influence of the CPhosILs ratio CPhosIL/substrate was studied. Results are summarized in Table 4 and best conditions were met with 0.5 eq. of CPhosIL 9. Indeed, good conversion of isophore was observed with high selectivity for trimethylcyclohexanone (TMCH) and with an important ee of 40% in favour of the isomer S. It also proved that CPhosILs were suitable solvents to induce good conversion, high selectivity and good ee for such mild process compared to other harder processes (Scheme 5).41
| Equivalent of 9 | Conv. (%) | Select. (%) | ee (%) |
|---|---|---|---|
| 1 | 7 | 93 | 32 (S) |
| 0.5 | 60 | 95 | 40 (S) |
 | ||
| Scheme 5 Hydrogenation of isophorone in CPhosIL 9. | ||
Recycling of the catalytic system led unfortunately to by-products as for example trimethylcyclohexanol by reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond (Scheme 6).
O bond (Scheme 6).
 | ||
| Scheme 6 Hydrogenation of isophorone in CPhosIL 9/solvent. | ||
Next, to envisage the recycling of the catalytic species, we tested several polar co-solvents which could facilitate the adsorption of the substrate on the catalyst and also participate to the solubilization of the hydrogen (H2) in the medium42 (Table 5).
| Solvent | Conv. (%) | Select. (%) | ee (%) |
|---|---|---|---|
| H2O | 96 | 95 | 5 |
| i-PrOH | 100 | 90 | 37 (S) |
| MeOH | 70 | 98 | 6 |
| H2O/MeOH (1/1) | 52 | 95 | 13 |
| Acetonitrile | 54 | 96 | 34 (S) |
| i-PrOH/H2O (2/1) | 17 | 67 | 11 |
| i-PrOH/H2O (3/1) | 43 | 93 | 2 |
The best ee (37%) was obtained using 9/i-PrOH as solvents. It also gave total conversion of isophorone with high selectivity for TMCH. Other co-solvents were tested as MeOH or mixtures H2O/MeOH or H2O/i-PrOH but led to lower conversions, selectivity and ee. Acetonitrile, a non-polar solvent gave similar ee than i-PrOH but with lower conversion, probably due to the weak solubility of H2. However, the recycling remained difficult as i-PrOH was miscible to diethylether. To conclude, no improvement of ee was observed using co-solvent.
The enantioselective hydrogenation was then performed on other α,β-unsaturated ketones using respectively TBP S-prolinate 9 or TBP trans-hydroxy-S-prolinate 11 as solvents (Scheme 7).
 | ||
| Scheme 7 Hydrogenation of α,β-unsaturated ketones with TBP S-prolinate 9 or TBP trans-hydroxy-S-prolinate 11 as solvents. | ||
We observed complete conversions and good diastereoisomeric excess (de) for the hydrogenation of R-carvone and R-pulegone in presence of 9. These results were excellent in comparison to literature data obtained with high pressure of H2 (5–10 bar) and/or high temperatures (40–100 °C).43 After a first run, the catalytic system had unfortunately lost some crucial activity and the use of 9 led generally to lower conversion for all ketones. The anion had an important effect on the reactivity. With PhosILs 11, both conversion and selectivity were significantly lowered; de was maintained with R-pulegone, but a drastic decreased was observed with R-carvone. The presence of the alcohol function on the chiral anion might cause steric hindrance or/and change H2 solubility of the CPhosILs. As a consequence, other substrate hydrogenations were pursued with only CPhosIL 9. Good conversion of β-ionone was measured with a good selectivity, the double carbon bond of the chain being preferentially hydrogenated compared to the cyclic one. Total conversion of trans-chalcone was observed with only formation of 3-phenylpropiophenone, as system was too soft to attack the aromatic double carbon bonds.
Experimental
All reagents were commercially available and used as received (TBPOH·30H2O, acids or diacids from Sigma-Aldrich). NMR spectra were recorded at 298 K at 500 MHz for 1H and at 63 MHz for 13C and 202 MHz for 31P on an Avance III Bruker spectrometer in CDCl3 as solvent with TMS or phosphoric acid as references.Concerning the quantification of the hydrogenation of linoleic acid, NMR spectroscopy was performed at 500 MHz with Bruker Avance III spectrometer equipped with a BBFO + probe and at 600 MHz with Bruker Avance III spectrometer equipped with a CPTCI cryoprobe. The sample was dissolved in CDCl3 or in MeOD-d4 and the resulting solution was placed in a 5 mm diameter NMR tube. 1H NMR spectra were taken with 30 degree pulse angle, 10 s relaxation delay and 16 scans.
IR spectra were recorded on a NICOLET IMPACT 400 spectrometer (KBr pellets or films).
Elemental analysis (C, H, N, S) were realized on a Flash EA-1112 Series.
GC analyses were recorded on a Hewlett–Packard HP-6890 gas chromatograph, fitted with DB-1 capillary column (25 m, 0.32 mm), a flame ionization detector and HP-3395 integrator under the following conditions: helium as vector gas (5 × 104 Pa), temperature of injector: 250 °C, temperature of the oven: isotherm 150 °C, 5 min, then 150–300 °C (10 °C min−1) and isotherm 300 °C, 5 min.
GC/MS analyses were recorded on a THERMOQUEST Draw GC on 2000 Series by using the techniques of chemical ionization under the following conditions: capillary column DB1 (length: 25 m, diameter: 0.32 mm), vector gas: helium (0.5 bar), temperature injector: 250 °C.
ee were determined with a β-cyclodextrines-based chiral gas chromatography column and by-products by GC/MS.
Thermogravimetric analyses coupled with a mass spectrometer were performed between 30 °C and 500 °C under a constant flow of dry argon (50 mL min−1) using a Simultaneous Thermal Analyzer STA 449C Jupiter from Netzsch, and a heating rate of 10 K min−1. The isothermal drift and sensitivity values are 0.6 μg h−1 and 0.1 μg, respectively. Alumina crucibles were loaded with 10–20 mg of sample powder.
The mass spectrometer was a quadrupole QMS 403 Aëolos® with a stainless steel capillary and a SEV detector (Channeltron). The counting time for mass spectrometer is of 20 ms per m/z values (scanning width: m/z = 10–150 amu) with a resting time of 1 s.
The DSC experiments were carried out on a Netzsch DSC 204F1 heat flux differential calorimeter at a heating/cooling rate of 10 K min−1 under a constant argon flow with 200 mL min−1. The crucibles were loaded with 10–20 mg of sample powder/liquid. Samples were weighed in aluminium sample pans covered with a pierced lid. An empty aluminium sample pan with a pierced lid was used as a reference.
The viscosities measurements in cP (or mPa s) were performed with a Brookfield LV-DVII + PRO viscometer using a CP51 cone spindle. The instrument was connected to a HUBER-ministat circulation-type thermo-regulated water bath, and measures were realized between 298.15 and 353.15 K. The repeatability of the viscometer was of 0.20% with an uncertainty in the viscosity measurements of 1.00% of the full scale range, declared by the manufacturer.
General procedure for the synthesis of PhosILs 1–3
General procedure for hydrogenation
Recycling experiments. After a short vacuum/argon sequence (three times), the substrate (2 mmol, 1 equiv.) was added, and the mixture was stirred vigorously for 18 h at room temperature under a hydrogen atmosphere (gas bag). Product isolation and characterization is the same as described above.
Conclusions
Phosphonium ionic liquids (PhosILs) with natural organic acid derived anions (L-lactate, L-tartrate, malonate, succinate, L-malate, pyruvate, D-glucuronate, D-galacturonate, S-prolinate, p-coumarate and ferulate) were easily prepared from tetrabutylphosphonium hydroxide and an excess of the corresponding acid with good yields. Their characterization was realized through routine NMR, IR, elemental analysis, viscosity, and ATG techniques. These PhosILs as co-solvents showed good performance, recyclability and better efficiency than commercial ILs in catalytic selective hydrogenation of 1,5-cyclooctadiene (1,5-COD) into cyclooctene at room temperature under atmospheric H2 pressure. Interesting extension of this hydrogenation process has been performed on unsaturated fatty acids, like linoleic acid. Fine NMR investigations proved the performance essentially concerning the selectivity of the catalytic system towards mono-unsaturated acids in the presence of PhosILs under mild conditions. Furthermore, enantioselective hydrogenation of α,β-unsaturated ketones in mild conditions was performed in pure chiral PhosILs. Very good conversion of substrates, combined with high selectivity and good enantiomeric or diastereoisomeric excesses revealed the high potentialities of our PhosILs for such processes compared to literature data.On-going investigations will deal with the evaluation of the biodegradability and/or (eco)toxicity of these new PhosILs in order to used them in other catalytic processes or extraction procedures.
Acknowledgements
This work was supported by the Fondation du Site Paris Reims (post doctoral fellowship for Nadège Ferlin) and the FEDER for material funds. We also deliver our many thanks and gratitude to the Tunisian Ministry of Education and Research for financial support for the cotutoring PhD of Safa Hayouni.References
- (a) J. D. Holbrey and K. R. Seddon, Clean Products and Processes, 1999, 1, 223–236 Search PubMed; (b) P. Wassercheid and T. Welton, Ionic liquids in synthesis, Wiley–VCH Weinheim, 2nd edn, 2008 Search PubMed.
- (a) C. M. Gordon, Appl. Catal., A, 2001, 222, 101–117 CrossRef CAS; (b) J. G. Huddleston, A. E. Visser, W. M. Reichert, H. D. Willauer, G. A. Broker and R. D. Rogers, Green Chem., 2001, 3, 156–164 RSC; (c) K. R. Seddon, J. Chem. Technol. Biotechnol., 1997, 68, 351–356 CrossRef CAS; (d) K. R. Seddon, A. Stark and M. J. Torres, Pure Appl. Chem., 2000, 72, 2275–2287 CrossRef CAS; (e) R. Sheldon, Chem. Commun., 2001, 2399–2407 RSC; (f) T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed; (g) P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed. Engl., 2000, 39, 3772–3789 CrossRef CAS; (h) D. Zhao, M. Wu, Y. Kou and E. Min, Catal. Today, 2002, 74, 157–189 CrossRef CAS; (i) J. Dupont, C. S. Consorti and J. Spencer, J. Braz. Chem. Soc., 2000, 11, 337–344 CAS; (j) C. J. Bradaric, A. Downard, C. Kennedy, A. J. Robertson and Y. Zhou, Strem Chem., 2003, 1, 1–333 Search PubMed.
- M. M. Rahut, H. A. Currier, A. M. Semsel and V. P. Wystrach, J. Org. Chem., 1961, 26, 5138–5145 CrossRef.
- A. J. Robertson, WO0187900, 2001, (Cytec Industries).
- N. Karodia, S. Guise, C. Newlands and J. Andersen, Chem. Commun., 1998, 2341–2342 RSC.
- (a) S. Bose, A. B. Wijeratne, A. Thite, G. A. Kraus, D. W. Armstrong and J. W. Petrich, J. Phys. Chem. B, 2009, 113, 10825–10829 CrossRef CAS PubMed; (b) X. Chen, X. Li, A. Hua and F. Wang, Tetrahedron: Asymmetry, 2008, 19, 1–14 CrossRef CAS.
- U. A. Rana, R. Vijayaraghavan, M. Walther, J. Sun, A. A. J. Torriero, M. Forsyth and D. R. Mac Farlane, Chem. Commun., 2011, 47, 11612–11614 RSC.
- S. I. Lall-Ramnarine, J. A. Mukhlall, J. F. Wishart, R. R. Engel, A. R. Romeo, M. Gohdo, S. Ramati, M. Berman and S. N. Suarez, Beilstein J. Org. Chem., 2014, 10, 271–275 CrossRef PubMed.
- D. Coleman and N. Gathergood, Chem. Soc. Rev., 2010, 39, 600–637 RSC.
- (a) P. Wasserscheid, A. Bösmann and C. Bolm, Chem. Commun., 2002, 3, 200–201 RSC; (b) H. Clavier, L. Boulanger, N. Audic, L. Toupet, M. Mauduit and J. C. Guillemin, Chem. Commun., 2004, 1224–1225 RSC; (c) L. González, B. Altava, M. Bolte, M. I. Burguete, E. García-Verdugo and S. V. Luis, Eur. J. Org. Chem., 2012, 26, 4996–5009 CrossRef; (d) W. Bao, Z. Wang and Y. Li, J. Org. Chem., 2003, 68(2), 591–593 CrossRef CAS PubMed; (e) J. C. Plaquevent, J. Levillain, F. Guillen, C. Malhiac and A. C. Gaumont, Chem. Rev., 2008, 108(12), 5035–5060 CrossRef CAS PubMed; (f) S. Luo, X. Mi, L. Zhang, S. Liu, H. Xu and J. P. Cheng, Angew. Chem., Int. Ed., 2006, 45, 3093–3097 CrossRef CAS PubMed; (g) D. Coleman, M. Špulák, M. T. Garcia and N. Gathergood, Green Chem., 2012, 14, 1350–1356 RSC; (h) A. Haiß, A. Jordan, J. Westphal, E. E. Logunova, N. Gathergood and K. Kümmerer, Green Chem., 2016, 18, 4361–4373 RSC; (i) A. Jordan, A. Haiß, M. Špulák, Y. Karpichev, K. Kümmerer and N. Gathergood, Green Chem., 2016, 18, 4374–4392 RSC; (j) Y. De Gaetano, J. Hubert, A. Mohamadou, S. Boudesocque, R. Plantier-Royon, J. H. Renault and L. Dupont, Chem. Eng. J., 2016, 285, 596–604 CrossRef CAS; (k) J. Pernak, M. Niemczak, L. Chrzanowski, L. Ławniczak, P. Fochtman, K. Marcinkowska and T. Praczyk, Chem. Eng. J., 2016, 22, 12012–12021 CAS.
- J. Pernak, F. Stefaniak and J. Weglewski, Eur. J. Org. Chem., 2005, 4, 650–652 CrossRef.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398–2399 CrossRef CAS PubMed.
- J. Kagimoto, K. Fukumoto and H. Ohno, Chem. Commun., 2006, 2254–2256 RSC.
- F. Atefi, M. T. Garcia, R. D. Singer and P. J. Scammells, Green Chem., 2009, 11, 1595–1604 RSC.
- (a) N. Ferlin, M. Courty, S. Gatard, M. Spulak, B. Quilty, I. Beadham, M. Ghavre, N. Gathergood and S. Bouquillon, Tetrahedron, 2013, 69, 6510–6518 CrossRef; (b) N. Ferlin, M. Courty, S. Gatard, M. Pour, B. Quilty, I. Beadham, M. Ghavre, N. Gathergood and S. Bouquillon, RSC Adv., 2013, 3, 26241–26251 RSC; (c) S. Hayouni, N. Ferlin and S. Bouquillon, Hydrogenation Catalysis in biobased Ionic Liquids, InTech - open science | open minds, just accepted.
- (a) D. Zhao, P. J. Dyson, G. Laurenczy and J. S. McIndoe, J. Mol. Catal. A: Chem., 2004, 214, 19–25 CrossRef CAS; (b) U. Kernchen, B. Etzold, W. Korth and A. Jess, Chem. Eng. Technol., 2007, 30, 985–994 CrossRef CAS; (c) J. Dupont, P. A. Z. Suarez, A. P. Umpierre and R. F. de Souza, J. Braz. Chem. Soc., 2000, 11, 293–297 CrossRef CAS.
- (a) Y. Chauvin, L. Mussmann and H. Olivier, Angew. Chem., Int. Ed., 1995, 34, 2698–2700 CrossRef CAS; (b) A. Z. P. Suarez, E. L. J. Dullius, S. Einloft, F. R. De Souza and J. Dupont, Polyhedron, 1996, 15, 217–1219 CrossRef.
- Green chemistry and catalysis, ed. R. A. Sheldon, I. Arends and U. Hanefeld, John Wiley & Sons, 2007, p. 448 Search PubMed.
- M. Ghavre, S. Morrissey and N. Gathergood, Hydrogenation in Ionic Liquids, Ionic Liquids: Applications and Perspectives, ed. A. Kokorin, InTech, 2011, pp. 331–392 Search PubMed.
- (a) S. C. Mhadgut, M. Török, S. Dasgupta and B. Török, Catal. Lett., 2008, 123, 156 CrossRef CAS; (b) N. Győrffy, A. Tungler and M. Fodor, J. Catal., 2010, 270, 2 CrossRef; (c) S. C. Mhadgut, M. Török, J. Esquibel and B. Török, J. Catal., 2006, 238, 441 CrossRef CAS; (d) S. C. Mhadgut, I. Bucsi, M. Török and B. Török, Chem. Commun., 2004, 984 RSC; (e) S. Li, C. Chen, E. Zhan, S.-B. Liu and W. Shen, J. Mol. Catal. A: Chem., 2009, 304, 88 CrossRef CAS; (f) S. Li, E. Zhan, Y. Li, Y. Xu and W. Shen, Catal. Today, 2008, 131, 347 CrossRef CAS; (g) S. C. Mhadgut, I. Bucsi, M. Török, S. Dasgupta and B. Török, Catal. Lett., 2008, 123, 156 CrossRef CAS; (h) M. Fodor, A. Tungler and L. Vida, Catal. Today, 2009, 140, 58 CrossRef CAS; (i) S. Li, E. Zhan, Y. Li, Y. Xu and W. Shen, Catal. Commun., 2007, 8, 1239 CrossRef.
- (a) M. Fodor, A. Tungler and L. Vida, Catal. Today, 2009, 140, 58 CrossRef CAS; (b) M. Fodor, A. Tungler and L. Vida, React. Kinet. Catal. Lett., 2007, 90, 413 CrossRef CAS; (c) É. Sípos, A. Tungler and I. Bitter, React. Kinet. Catal. Lett., 2003, 79, 101 CrossRef; (d) É. Sípos, A. Tungler and I. Bitter, J. Mol. Catal. A: Chem., 2003, 198, 167 CrossRef; (e) A. Tungler, T. Máthé, T. Tarnai, K. Fodor, G. Tóth, J. Kajtár, I. Kolossváry, B. Herényi and R. A. Sheldon, Tetrahedron: Asymmetry, 1995, 6, 2395 CrossRef CAS; (f) N. Győrffy and A. Tungler, J. Mol. Catal. A: Chem., 2011, 336, 72 CrossRef; (g) N. Győrffy and A. Tungler, J. Mol. Catal. A: Chem., 2011, 336, 72 CrossRef; (h) G. Fogassy, A. Tungler, A. Lévai and G. Tóth, J. Mol. Catal. A: Chem., 2002, 179, 101 CrossRef CAS; (i) É. Sípos, A. Tungler and I. Bitter, J. Mol. Catal. A: Chem., 2003, 198, 167 CrossRef; (j) É. Sípos, A. Tungler, I. Bitter and M. Kubinyi, J. Mol. Catal. A: Chem., 2002, 186, 187 CrossRef; (k) G. Farkas, K. Fodor, A. Tungler, T. Máthé, G. Tóth and R. A. Sheldon, J. Mol. Catal. A: Chem., 1999, 138, 123 CrossRef CAS.
- (a) É. Sípos, G. Farkas, A. Tungler and J. L. Figueiredo, J. Mol. Catal. A: Chem., 2002, 179, 107 CrossRef; (b) G. Farkas, É. Sípos, A. Tungler, A. Sárkány and J. L. Figueiredo, J. Mol. Catal. A: Chem., 2001, 170, 101 CrossRef CAS; (c) A. Tungler, Y. Nitta, K. Fodor, G. Farkas and T. Máthé, J. Mol. Catal. A: Chem., 1999, 149, 135 CrossRef CAS.
- H.-U. Funk, G. Senft and H.-D. Quest, Eur. Patent EP 1318130A1, 2003.
- H. Yoshiyuki, Jpn. Kokai Tokkyo Koho JP 63057545, 2000.
- (a) M. Pisarek, M. Lukaszewski, P. Winiarek, P. Kedzierzawski and M. Janik-Czachor, Catal. Commun., 2008, 10, 213 CrossRef CAS; (b) M. Pisarek, M. Lukaszewski, P. Winiarek, P. Kedzierzawski and M. Janik-Czachor, Mater. Chem. Phys., 2009, 114, 774 CrossRef CAS.
- T. Sato, S. Watanabe, H. Kiuchi, S. Oi and Y. Inoue, Tetrahedron Lett., 2006, 47, 7703 CrossRef CAS.
- T. Sato, C. V. Rode, O. Sato and M. Shirai, Appl. Catal., B, 2004, 49, 181 CrossRef CAS.
- C. I. Melo, R. Bogel-Łukasik, M. Gomes da Silva and E. Bogel-Łukasik, Green Chem., 2011, 13, 2825 RSC.
- (a) A. Strádi, M. Molnár, M. Óvári, G. Dibó, F. U. Richter and L. Mika, Green Chem., 2013, 15(7), 1857–1862 RSC; (b) A. Strádi, M. Molnár, P. Szakál, G. Dibó, D. Gáspár and L. T. Mika, RSC Adv., 2015, 5(89), 72529–72535 RSC.
- V. Pandarus, G. Gingras, F. Béland, R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2012, 16, 1307–1311 CrossRef CAS.
- (a) S. Breton, A. Brisach-Wittmeyer, J. J. Rios Martin, M. L. Camacho, A. Lasia and H. Ménard, Int. J. Electrochem., 2011, 1–9 CrossRef; (b) K. R. Kahsar, D. K. Schwartz and J. W. Medlin, ACS Catal., 2013, 3, 2041–2044 CrossRef CAS.
- (a) F. Endres and S. Zein El Abedin, Phys. Chem. Chem. Phys., 2006, 8, 2101–2116 RSC; (b) J. Tindale, C. Na, M. Jennings and P. Ragogna, Can. J. Chem., 2007, 85(10), 660–667 CrossRef CAS; (c) F. Endres and S. Zein El Abedin, Phys. Chem. Chem. Phys., 2006, 8, 2101–2116 RSC.
- (a) M. C. Gálvez, C. G. Barroso and J. A. Pérez-Bustamante, Z. Lebensm.-Unters. Forsch., 1994, 199, 29–31 CrossRef; (b) Z. Quinde-Axtell and B. K. Zory, J. Agric. Food Chem., 2006, 54(26), 9978–9984 CrossRef CAS PubMed.
- N. Kumar and V. Pruthi, Biotechnol. Rep., 2014, 4, 86–93 CrossRef.
- V. Di Castro, C. Furlani, C. Fragale, M. Gargano, M. Rossi and N. Ravasio, Gazz. Chim. Ital., 1987, 117, 43–50 CAS.
- (a) D. Camper, C. Becker, C. Koval and R. Noble, Ind. Eng. Chem. Res., 2006, 45, 445–450 CrossRef CAS; (b) P. J. Dyson, G. Laurenczy, C. A. Ohlin, J. Vallance and T. Welton, Chem. Commun., 2003, 2418–2419 RSC.
- K. Yamamoto, A. Kinshita and A. Shibahara, J. Chromatogr., 2008, 1182, 132–135 CrossRef CAS PubMed.
- J. K. G. Kramer, C. Cruz-Hernandez and J. Zhou, Eur. J. Lipid Sci. Technol., 2001, 103, 594–632 CrossRef CAS.
- (a) R. Sacchi, F. Addeo and L. Paolillo, Magn. Reson. Chem., 1997, 35, S133–S145 CrossRef CAS; (b) Y. Miyake, K. Yokomizo and N. Matsuzaki, J. Am. Oil Chem. Soc., 1998, 75, 1091–1094 CrossRef CAS.
- M. S. Carvalho, R. A. Lacerda, J. P. Leão, J. D. Scholten, B. A. Neto and P. A. Z. Suarez, Catal. Sci. Technol., 2011, 1, 480–488 CAS.
- (a) M. Fodor, A. Tungler and L. Vida, Catal. Today, 2009, 140, 58–63 CrossRef CAS; (b) A. Tungler, J. Kajtar, T. Mathé, G. Toth, E. Fogassy and J. Petro, Catal. Today, 1989, 5, 159–171 CrossRef CASA. Tungler, T. Máthé, J. Petró and T. Tarnai, J. Mol. Catal. A: Chem., 1990, 61, 259–267 CrossRef CAS; (c) E. Sipos, A. Tungler, I. Bitter and M. Kubinyi, J. Mol. Catal. A: Chem., 2002, 186, 187–192 CrossRef CAS; (d) E. Sipos, A. Tungler and I. Bitter, J. Mol. Catal. A: Chem., 2003, 198, 167–173 CrossRef CAS; (e) E. Sipos, G. Fogassy, A. Tungler, P. V. Samant and J. L. Figueiredo, J. Mol. Catal. A: Chem., 2004, 212, 245–250 CrossRef CAS.
- (a) S. Kishida and S. Teranishi, J. Catal., 1968, 12, 90–96 CrossRef CAS; (b) Y. Sun, R. N. Landau, J. Wang, C. Le Blond and D. G. Blackmond, J. Am. Chem. Soc., 1996, 118, 1348–1353 CrossRef CAS; (c) P. Dyson, G. Laurenczy, A. Ohlin, J. Vallance and T. Welton, Chem. Commun., 2003, 2418–2419 RSC.
- F. A. Khan, A. Vallat and G. Süss-Fink, J. Mol. Catal. A: Chem., 2012, 355, 168–173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23056c |
| This journal is © The Royal Society of Chemistry 2016 |