
Surface modification of promising cerium oxide nanoparticles for nanomedicine applications
Himansu Sekhar Nanda†
*
Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, Saudi Arabia
First published on 14th November 2016
Abstract
Cerium oxide nanoparticles (CNPs) or nanoceria have emerged as a potential nanomedicine for the treatment of several diseases such as cancer. CNPs have a natural tendency to aggregate or agglomerate in their bare state, which leads to sedimentation in a biological environment. Since the natural biological environment is essentially aqueous, nanoparticle surface modification using suitable biocompatible hydrophilic chemical moieties is highly desirable to create effective aqueous dispersions. In this report, (6-{2-[2-(2-methoxy-ethoxy)-ethoxy]-ethoxy}-hexyl)triethoxysilane was used as a functional, biocompatible organosilane to modify the surface of CNPs to produce promising nanoparticles which open substantial therapeutic avenues. The surface modified nanoparticles were produced in situ via an ammonia-induced ethylene glycol-assisted precipitation method and were characterized using complimentary characterization techniques. The interaction between the functional moiety and the nanoparticle was studied using powerful cross polarization/magic angle sample spinning solid state nuclear magnetic resonance spectroscopy. The surface-modified nanoparticles were extremely small and demonstrated a significant improvement in aqueous dispersibility. Moreover, the existence of a strong ionic coordination between the functional moiety and the surface of the nanoparticle was realised, indicating that the surface modified nanoceria are stable and that the nanoparticles should demonstrate an enhanced circulation time in a biological environment. The surface modification approach should be promising for the production of CNPs for nanomedicine applications.
1. Introduction
Introduction of nanotechnology to biology and medicine has resulted in an unprecedented number of nanomaterials for therapeutic applications.1 For example, carrier and non-carrier-based nanoparticles have been investigated for their potential applications in nanomedicine.2 Cerium (Ce) oxide (CeO2) nanoparticles (CNPs) or nanoceria as non-carrier metal oxide nanoparticles have shown its profound use in biology and nanomedicine.3–5 Because of the auto-regeneration of Ce valence states (Ce3+ and Ce4+), these nanoparticles can act as both a pro-oxidant and an antioxidant.4 The biological action of CNPs depends on the ratio of dual oxidation states (Ce3+ to Ce4+). Doping of CNPs with other transition metal ions has been reported as one method for altering and controlling nanoparticle properties and is important for tuning the nanoparticle properties.6 CNPs in the +3 oxidation state scavenge superoxide radical anions in living cells, mimicking the activity of superoxide dismutase (SOD). In the +4 oxidation state, CNPs facilitate the decomposition of H2O2 to O2 and H2O, mimicking the role of a catalase.4 CNPs scavenge reactive ion species, such as reactive oxygen species (ROS) and reactive nitrogen species (RNS), and have potential therapeutic applications against several oxidative stress related disorders.5 Furthermore, CNPs have shown inhibitory effects on cancer progression and are toxic to tumor cells and non-toxic to stromal cells. They have also been investigated as a potential nanomedicine for cancer treatment.4,5,7,8 The therapeutic performance of these nanoparticles depends on optimal and effective biological dispersion throughout the duration of the treatment and remains a major obstacle to their clinical application.5–9Due to the large number of surface defects, CNPs have a natural tendency to aggregate in their bare state if they are not functionalized.9 The aggregation results in quick sedimentation, deposition in the reticuloendothelial system (RES) and toxicity.10 Non-functionalized CNPs in a high ionic medium, such as phosphate buffer saline (PBS) and cell culture medium (DMEM), have been shown to aggregate and form larger particles within a few hours.9–11 The sedimentation is faster as the nanoparticle concentration increases. In order to overcome these drawbacks, the synthesis of biocompatible CNPs in pure water, citrate, cyclodextrin, polymers and with surface modification using a few functionalizing ligand moieties such as polyethylene glycol and heparin have been reported.7,12–14 Most of the above mentioned protecting agents were either found to be readily washed off (due to the weak chemical interaction between the nanoparticle and ligand functionalities) or reduce the catalytic activity (due to their large size and high coating density) upon exposure to physiological conditions.9,15
Therefore, it is important to search for new functionalizing moieties as well as novel in situ surface engineering strategies for production of robust CNPs for potential nanomedicine applications. Furthermore, the physicochemical characterization of the surface modified CNPs is a pre-requisite for an adequate assessment of their biological application. The use of organosilanes has been reported for engineering the surface of magnetic and silica nanoparticles, but not for CNPs.16,17 Surface engineering of the CNPs could be possible via a silanization process under defined and optimized reaction conditions.18 Possible functionalization of CNPs with hydrophilic biocompatible functional organosilane moieties could yield CNPs with improved aqueous dispersibility, which might address the issues related to clinical applications.18,19 In this study, we report the use of (6-{2-[2-(2-methoxy-ethoxy)-ethoxy]-ethoxy}-hexyl)triethoxysilane (MEEETES) as a hydrophilic biocompatible organosilane to engineer the surface of CNPs produced via an ammonia-induced ethylene glycol-assisted precipitation method. The surface engineering was carried out in situ using a single and two-step synthetic procedure. The surface modified CNPs were characterized using complementary techniques including powerful 13C cross polarization magic angle sample spinning solid state-nuclear magnetic resonance to obtain a better understanding of the altered CNPs.
2. Experimental
2.1 Preparation and surface engineering of cerium oxide nanoparticles
The unmodified and surface modified CNPs were synthesized by an ammonia-induced ethylene glycol (EG)-assisted precipitation method.18 Briefly, 7.8 mL (0.12 mol) of EG (99%, Sigma Aldrich, St. Louis, MO, USA) were added to 92.2 mL of deionized water (Milli-Q® Direct Water Purification System, Merck Millipore, Billerica, MA, USA) in a 250 mL two neck round bottom distillation flask at 50 °C in a silicone bath reflux condenser system with constant magnetic stirring at 350 rpm. Cerium(III) nitrate hexahydrate (5.16 g, 0.012 mol) (Ce(NO3)3·6H2O) (99.99%, Sigma Aldrich, St. Louis, MO, USA) was added into the EG/H2O solution at an EG to Ce3+ molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. After complete dissolution of Ce(NO3)3·6H2O, 5 mL of aqueous ammonia (NH4OH) (29.44%, Fisher Scientific, Pittsburgh, PA, USA) were added to the solution and the pH was adjusted to 9.6. The solution was kept under constant stirring at 750 rpm and was allowed to react until the colour of the solution became yellow (as a confirmation of CNP formation). Once CNP formation was confirmed, the stirring was stopped, and the entire reaction volume was subjected to vacuum filtration using standard Whatman (Ø = 110 mm and grade 589/3) filter paper over a Buchner funnel. The filtered nanomaterials were subjected to an alternate wash with ethanol and deionized water (6 times each) and were dried overnight in a fume hood. The dried nanomaterials were crushed and ground in a ceramic mortar using a pestle and then freeze-dried using a freeze-drier (Labconco Corporation, Kansas City, MO, USA). The freeze-dried nanoparticles were further crushed and ground using the ceramic mortar and pestle to ensure the production of fine, powdered CNPs. The prepared, unmodified CNPs were used as a control to compare against the surface modified CNPs.
:1. After complete dissolution of Ce(NO3)3·6H2O, 5 mL of aqueous ammonia (NH4OH) (29.44%, Fisher Scientific, Pittsburgh, PA, USA) were added to the solution and the pH was adjusted to 9.6. The solution was kept under constant stirring at 750 rpm and was allowed to react until the colour of the solution became yellow (as a confirmation of CNP formation). Once CNP formation was confirmed, the stirring was stopped, and the entire reaction volume was subjected to vacuum filtration using standard Whatman (Ø = 110 mm and grade 589/3) filter paper over a Buchner funnel. The filtered nanomaterials were subjected to an alternate wash with ethanol and deionized water (6 times each) and were dried overnight in a fume hood. The dried nanomaterials were crushed and ground in a ceramic mortar using a pestle and then freeze-dried using a freeze-drier (Labconco Corporation, Kansas City, MO, USA). The freeze-dried nanoparticles were further crushed and ground using the ceramic mortar and pestle to ensure the production of fine, powdered CNPs. The prepared, unmodified CNPs were used as a control to compare against the surface modified CNPs.
Surface modified CNPs were produced by functionalization of the surface of the CNPs with MEEETES moieties and were synthesized in situ using a similar synthetic procedure. For production of MEEETES functionalized CNPs (MEEETES-CNPs), the synthetic procedure for unmodified CNPs was followed up until the formation of the CNPs. The surface engineering was an intermediate step in the reaction. Instead of the CNP recovery used in the former synthetic procedure, 400 μL of MEEETES (20 mM) (SiKEMIA, Montpellier, France) was added to the 100 mL reaction volume under an inert atmosphere and allowed to react. The reaction was carried out until the solution turned a milky yellow (an indication of the formation of surface modified CNPs). Once the formation was confirmed, the stirring was stopped and the MEEETES-CNPs were recovered via centrifugation (17000 rpm, 10 min, 4 °C) using a high speed refrigerated centrifuge (Avanti J-26XP, Coulter Inc., Brea, CA, USA). The MEEETES-CNPs were washed using a mixture of ethanol and deionized H2O (1:1 volume ratio) in a similar centrifugation procedure under identical centrifugation conditions. The pellets in the centrifuge tubes were dispersed in deionized water and freeze-dried. The freeze-dried nanoparticles were crushed and ground using the ceramic mortar and pestle to ensure the production of fine and dried MEEETES-CNPs.
2.2 Characterization
2.3 Cross polarization magic angle sample spinning 13C solid state nuclear magnetic resonance spectroscopy
Cross polarization magic angle sample spinning (CP-MAS) solid state 13C nuclear magnetic resonance (13C solid state-NMR) on CNPs and MEEETES-CNPs was performed on a Bruker Avance III 400 MHz spectrometer (Bruker Corporation, Billerica, MA, USA) equipped with a triple-resonance 4 mm Bruker MAS probe. 13C CP-MAS NMR spectra were recorded at a resonance frequency of 100.622 MHz under a 14 kHz spinning rate. The temperature for all experiments was 298 K. The cross-polarization contact time was set to 2 ms employing ramp 100 for variable amplitude CP. Bruker Topspin 3.0 software (TopSpin®, Bruker Corporation, Billerica, MA, USA) was used for data collection and spectral analysis.3. Results and discussion
The surface modification of CNP was carried out in two distinct steps in a single synthetic procedure. The steps were ionic coordination of the –OH groups (equivalent to the hydroxylation in the silica nanoparticles) to the CNP surface using EG and the subsequent competitive substitution by reactive, stable and preferred MEEETES with the previously coordinated –OH groups. Fig. 1 shows the TEM and HR-TEM images of the modified and unmodified nanoparticles. The microscopic images revealed the production of extremely small polyhedral shape nanoparticles 5–10 nm in size. No significant size difference was observed between the CNPs and MEEETES-CNPs, indicating that the surface modification did not affect the individual nanoparticle size. The results demonstrated that the functional organosilane moieties were extremely small. TEM images showed significant differences in the dispersion of the unmodified and modified CNPs. The MEEETES-CNPs had a more homogeneous dispersion and less aggregation than the unmodified CNPs.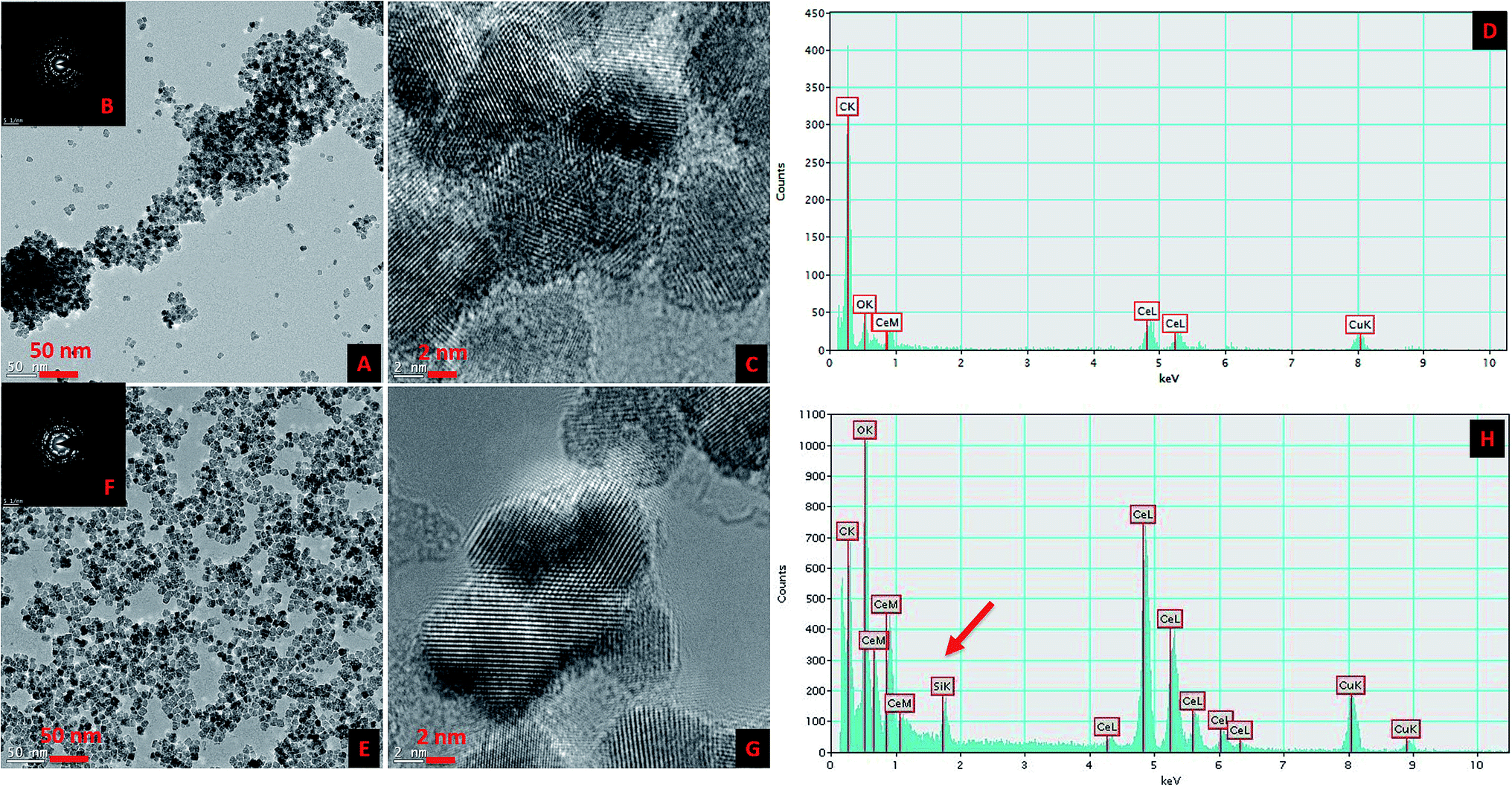 | ||
| Fig. 1 Transmission electron microscopic (TEM) images (A and E), high resolution TEM (HR-TEM) images (C and G), selected area electron diffraction (SAED) (B and F) and energy-dispersive X-ray analysis (EDXA) (D and H) of unmodified CNPs (A–D) and surface modified CNPs (MEEETES-CNPs) (E–H). The red arrow indicates the presence of the additional element “Si” in the EDXA of MEEETES-CNPs, which is absent in the unmodified CNPs. | ||
The information obtained from TEM was further validated using DLS measurements (Fig. 2). DLS cannot discriminate between inorganic and organic materials and measures the overall nanoparticle size, i.e., the hydrodynamic diameter of the nanoparticles in their best possible aggregated state.20 The aggregate size or hydrodynamic diameter (d nm) was found to have a single significant peak for the average particle size (mean z-average) at 200.6 ± 0.5 nm for the CNPs and 96.8 ± 2.2 nm for the MEEETES-CNPs (Fig. 2B). The results indicated that the nanoparticle aggregation was significantly reduced after the surface modification process and that the MEEETES-CNPs have better aqueous dispersibility than the CNPs since the measurement of DLS was carried out in deionized water.
 | ||
| Fig. 2 Powdered X-ray diffraction (XRD) patterns (A) and hydrodynamic size distribution (B) of the CNPs and MEEETES-CNPs. | ||
Fig. 3 shows the zeta potential distribution of CNPs and MEEETES-CNPs. The measured zeta potential values of CNPs and MEEETES-CNPs were −32.0 ± 2.1 mV and −42.3 ± 3.2 mV, respectively. The zeta potential is related to the charge on the surface of the nanoparticle and is an essential indicator of the stability of the nanoparticles in a colloidal system.21 In principle, if all the particles in a suspension have a large negative or positive zeta potential, they will repel each other and there is no tendency to flocculate or aggregate. However, if the particles have low zeta potential values, there is no force to prevent the particles from coming together and there is a higher tendency to flocculate. The general dividing line between stable and unstable suspensions is at either +30 mV or −30 mV. Particles with zeta potential values more positive than +30 mV or more negative than −30 mV are normally considered stable, and the stability of the nanoparticles increases with increasing corresponding positive or negative values of zeta. Based on the zeta potential values, the MEEETES-CNPs showed a greater stability than the CNPs in a colloidal state since the medium for the measurement was deionized water. The higher colloidal stability of the nanoparticles is extremely important for the safe use of nanoparticles in nanomedicine applications.21 The improved water dispersibility and higher colloidal stability of the MEEETES-CNPs could be due to the surface hydrophilicity in combination with the electrostatic and steric repulsion among the nanoparticles after surface modification.19
 | ||
| Fig. 3 Zeta potential value distribution of CNPs (A) and MEEETES-CNPs (B) in deionized water. | ||
The crystallinity and ordered structure of the nanoparticles were verified from the SAED, HR-TEM and XRD measurements. The SAED of both nanoparticles showed ring-like patterns (Fig. 1B and F). The images revealed high crystallinity and ordered structure of the nanoparticles' lattice planes. Both SAED and HR-TEM images (Fig. 1C and G) demonstrated that the lattice fringes can be clearly observed for both nanoparticles. The powdered XRD patterns of the CNPs and MEEETES-CNPs indicated that the diffraction peaks of the nanoparticles were indexed as the CeO2 phase (Fig. 2A). The characteristic diffraction peaks marked respectively by their indices (111), (200), (220), (311), (222), (400), (331) and (420) should be well indexed to the face centred cubic (FCC) fluorite structure of the available CNPs.22 More importantly, the characteristic peaks did not change (other than the intensity and a minor variation in width) after the surface modification process. Furthermore, no additional amorphous “halo” was observed in the XRD pattern of the MEEETES-CNPs. This indicated that the crystalline structure of the nanoparticles was unaltered after the surface modification. Moreover, the peaks were broad, which indicated the formation of very fine particles in the nanoscale regime and was in agreement with the results obtained from the TEM.
The EDXA analysis of CNPs and MEEETES-CNPs demonstrated the presence of additional “Si” in MEEETES-CNPs (Fig. 1D and H) as an indication of the presence of MEEETES in the surface modified CNPs. In order to obtain a better understanding of the surface modification, 13C CP-MAS solid state NMR was used to study the chemical composition and molecular structure of the surface modified CNPs. The NMR spectrum of the MEEETES-CNPs shows eight peaks that can be assigned to the different carbon atoms of the MEEETES moiety (Fig. 4). It was expected that the ethoxy groups of MEEETES would give two very strong, almost identical NMR signals at 18.2 ppm (![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) H3–CH2–O–) and at 58.4 ppm (CH3–H2–O–).23 The absence of those signals in the spectrum indicated that all the alkoxysilanes were bonded to the surface of the nanoparticles since the nanoparticles were washed thoroughly after the surface modification. All other signals were assigned according to published reports and are in agreement with the structure proposed.23,24 The signal at 14.08 ppm, corresponding to the (–SiH2–) group, is distinctly broader than the other signals. This might be because not all of the MEEETES molecules bonded with all three Si–O–CeO2 and some only bonded with two or one, namely R–Si(OH)(Si–O–)2 or R–Si(OH)2(Si–O–) (Fig. 5). However, this does not contradict the conclusion that all functional MEEETES moieties are strongly bonded to the surface of the nanoparticles.
H3–CH2–O–) and at 58.4 ppm (CH3–H2–O–).23 The absence of those signals in the spectrum indicated that all the alkoxysilanes were bonded to the surface of the nanoparticles since the nanoparticles were washed thoroughly after the surface modification. All other signals were assigned according to published reports and are in agreement with the structure proposed.23,24 The signal at 14.08 ppm, corresponding to the (–SiH2–) group, is distinctly broader than the other signals. This might be because not all of the MEEETES molecules bonded with all three Si–O–CeO2 and some only bonded with two or one, namely R–Si(OH)(Si–O–)2 or R–Si(OH)2(Si–O–) (Fig. 5). However, this does not contradict the conclusion that all functional MEEETES moieties are strongly bonded to the surface of the nanoparticles.
 | ||
| Fig. 4 The 13C CP-MAS spectrum of MEEETES-CNPs with annotated peaks and the chemical structure of MEEETES as a reference. | ||
 | ||
| Fig. 5 Schematic and possible mechanism of the two step synthetic procedure for the formation of MEEETES-CNPs and the possible chemical structure of the MEEETES-CNP. | ||
The mechanism of the MEEETES surface modification was proposed based on the bonding theory from the inner- and outer-sphere model of CNPs proposed by Eric Salazar-Sandoval Johansson.25 Fig. 5 shows the schematic of the possible mechanism of MEEETES-CNP formation. Because of the small size and high charge of the cerium cations, the outermost surface of the cerium cations of the CNPs prefer to coordinate through an ionic bond.25 CNPs are ionic nanocrystals. The successful ligands can only interact with the surface of the nanoparticles via ionic bonds.25 Therefore, any direct bonding extended from the CNP surface has to be essentially ionic. It is important to note that the surface chemistry of an ionic nanocrystal (e.g., a cerium oxide nanoparticle) is different than that of a covalent nanocrystal (e.g., a silica nanoparticle) and must be treated differently when proposing the mechanism for ligand mediated surface modification. This is a critical point of discussion in this study. The organosilane modification of the covalent nanocrystals, such as magnetic and silica nanoparticles, has already been reported and the mechanism of surface modification has been well demonstrated.16,17 However, the present study elucidates the mechanism for an organosilane modification of an ionic nanocrystal where the mechanism of silanization is different than other covalent nanocrystals reported in published studies. CNPs consist of a surface layer of Ce2O3 and a core CeO2.26 The aqueous stability of Ce2O3 (Ce3+) is greater than that of CeO2 (Ce4+).26 The hydroxyl (–OH) groups of the hydroxylated surface of the CNPs formed by the hydrolysis of an anion or the cerium cation (in the initial step of the synthesis) are coordinated ionically to the inner sphere of the CNP surface cations. The subsequent ligand exchange process (the 2nd step of the reaction) is a competitive ligand substitution, which indicates that a more stable and preferred ligand MEEETES has replaced the previously coordinated –OH groups to form a stable inner-sphere surface ionic complex of MEEETES-CNP.
4. Conclusion
For nanomedicine applications, nanoparticles need to be stable with respect to aggregation and dispersible in aqueous media. This report provides a generic approach to engineer the surface of cerium oxide nanoparticles with a small biocompatible organosilane moiety. The chelating ability of (6-{2-[2-(2-methoxy-ethoxy)-ethoxy]-ethoxy}-hexyl)triethoxysilane (MEEETES) was exploited for in situ modification of the CNP surface via an ammonia-induced ethylene glycol-assisted precipitation method. Furthermore, MEEETES has been shown to coordinate to the inner-sphere of the nanoparticle surface cations via a strong ionic interaction verified by a combination of experimental (powerful 13C CP-MAS solid state NMR) and theoretical (inner and outer sphere model of the nanoceria) studies. The knowledge gained during this investigation shows that small biocompatible organosilanes should be pursued as cerium oxide nanoparticle surface modifiers because they can form stable inner-sphere surface ionic complexes. The formation of strong and stable ionic complex between the functional moieties and the surface of nanoparticle indicates MEEETES moieties cannot be easily desorbed from the surfaces of the nanoparticles. The altered cerium oxide nanoparticles should demonstrate an enhanced stability and circulation time in natural biological environment and open promising therapeutic avenues in nanomedicine.Conflict of interests
The author declares that there is no conflict of interest regarding the publication of this research article.Acknowledgements
The author would like to acknowledge the financial support from King Abdullah University of Science and Technology (KAUST), Saudi Arabia.References
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther., 2008, 83, 761–769 CrossRef PubMed.
- Y. Liu, H. Miyoshi and M. Nakamura, Int. J. Cancer, 2007, 120, 2527–2537 CrossRef PubMed.
- C. Xu and X. Qu, NPG Asia Mater., 2014, 6, e90 CrossRef.
- M. S. Wason and J. Zhao, Am. J. Transl. Res., 2013, 5, 126–131 Search PubMed.
- Y. Gao, K. Chen, J.-l. Ma and F. Gao, OncoTargets Ther., 2014, 7, 835 CrossRef PubMed.
- J. C. Bear, P. D. McNaughter, P. Southern, P. O'Brien and C. W. Dunnill, Crystals, 2015, 5, 312–326 CrossRef.
- E. Alpaslan, H. Yazici, N. H. Golshan, K. S. Ziemer and T. J. Webster, ACS Biomater. Sci. Eng., 2015, 1, 1096–1103 CrossRef.
- A. Asati, S. Santra, C. Kaittanis, S. Nath and J. M. Perez, Angew. Chem., Int. Ed., 2009, 48, 2308–2312 CrossRef PubMed.
- A. Kumar, S. Das, P. Munusamy, W. Self, D. R. Baer, D. C. Sayle and S. Seal, Environ. Sci.: Nano, 2014, 1, 516–532 RSC.
- A. Asati, S. Santra, C. Kaittanis and J. M. Perez, ACS Nano, 2010, 4, 5321–5331 CrossRef PubMed.
- B. Chanteau, J. Fresnais and J.-F. Berret, Langmuir, 2009, 25, 9064–9070 CrossRef PubMed.
- A. Cimini, B. D'Angelo, S. Das, R. Gentile, E. Benedetti, V. Singh, A. M. Monaco, S. Santucci and S. Seal, Acta Biomater., 2012, 8, 2056–2067 CrossRef PubMed.
- A. Gupta, S. Das, C. J. Neal and S. Seal, J. Mater. Chem. B, 2016, 4, 3195–3202 RSC.
- J. M. Perez, A. Asati, S. Nath and C. Kaittanis, Small, 2008, 4, 552–556 CrossRef PubMed.
- W. J. Stark, Angew. Chem., Int. Ed., 2011, 50, 1242–1258 CrossRef PubMed.
- B. Feng, R. Hong, L. Wang, L. Guo, H. Li, J. Ding, Y. Zheng and D. Wei, Colloids Surf., A, 2008, 328, 52–59 CrossRef.
- V. Cauda, A. Schlossbauer and T. Bein, Microporous Mesoporous Mater., 2010, 132, 60–71 CrossRef.
- F. Caputo, PhD thesis, University of Rome Tor Vergata, 2014.
- R. De Palma, S. Peeters, M. J. Van Bael, H. Van den Rul, K. Bonroy, W. Laureyn, J. Mullens, G. Borghs and G. Maes, Chem. Mater., 2007, 19, 1821–1831 CrossRef.
- A. B. Salunkhe, V. M. Khot, N. D. Thorat, M. R. Phadatare, C. I. Sathish, D. S. Dhawale and S. H. Pawar, Appl. Surf. Sci., 2013, 264, 598–604 CrossRef.
- M. S. Darwish and I. Stibor, J. Dispersion Sci. Technol., 2016, 37, 1793–1798 CrossRef.
- S. Babu, R. Thanneeru, T. Inerbaev, R. Day, A. E. Masunov, A. Schulte and S. Seal, Nanotechnology, 2009, 20, 085713 CrossRef PubMed.
- E. Breitmaier and G. Bauer, 13C NMR spectroscopy: a working manual with exercises, CRC Press, Boca Raton, FL, 1984 Search PubMed.
- T. Sen and I. J. Bruce, Sci. Rep., 2012, 2, 564 Search PubMed.
- E. S.-S. Johansson, Ceria Nanoparticle Hybrid Materials: Interfacial Design and Structure Control, 2015 Search PubMed.
- A. Vincent, T. M. Inerbaev, S. Babu, A. S. Karakoti, W. T. Self, A. E. Masunov and S. Seal, Langmuir, 2010, 26, 7188–7198 CrossRef PubMed.
Footnote |
| † Present Address: School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Email: E-mail: hnanda@ntu.edu.sg; , binodinitifr@gmail.com |
| This journal is © The Royal Society of Chemistry 2016 |