
Functional characterizations of interactive recombinant PTEN–silica nanoparticles for potential biomedical applications†
Abstract
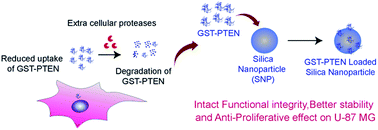
The complex architecture of recombinant proteins poses a major challenge in their formulation and delivery. Preservation of function and structural integrity is the fundamental prerequisite to achieve success in biomedical applications. Amid numerous formulations, maneuvering nanoparticles for optimal protein stabilization is attracting great interest. Herein, we provide a systematic characterization of stabilization of a tumor suppressor protein PTEN onto silica nanoparticles for biomedical applications. PTEN, a critical intracellular signaling protein is down regulated in several cancers leading to abnormal cell growth. The tumor suppression property of PTEN relies on its protein and particularly lipid phosphatase activity. Kinetic parameters Km and kcat/Km of purified GST tagged and untagged PTEN towards para-nitrophenylphosphate and phosphatidylinositol 3,4,5-trisphosphate (diC8) exhibited no significant difference suggesting that the GST tag does not hinder the enzymatic property of PTEN. Structural conformation of silica immobilized GST–PTEN was sustained as evident by circular dichroism measurement. Subsequently, GST–PTEN immobilized onto silica nanoparticles retained its phosphatase activity indicating successful stabilization. A protease protection assay indicated improved stability of the silica nanoparticles bound protein. The effect of GST–PTEN–silica nanoconjugate was evaluated on a PTEN null cell line U-87 MG, which revealed significant reduction in cell proliferation. Maintenance of the functional and structural framework of GST–PTEN immobilized silica nanoparticles along with its anti-proliferative role in glioblastoma cell lines boosts its potential application to combat PTEN deficient conditions.
 Please wait while we load your content...
Please wait while we load your content...

