
White light emission from quantum dot and a UV-visible emitting Pd-complex on its surface†
Abstract
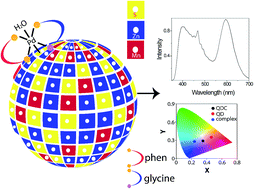
Near white light emission was achieved following surface functionalization of Mn-doped ZnS quantum dots (Qdots) by [Pd(glycine)(phenanthroline)]Cl complex with a proposed bonding of the complex through the dangling sulphide ion of the Qdot. Reaction between the complex ion and Qdot lead to the formation of quantum dot complex. Independent emissions in the UV-vis region from the complex and orange from the doped quantum dot lead to such color. Also, the material exhibited a dual decay mechanism comprising of fluorescence and phosphorescence pathways. The strategy described in the present report, with two independent emission channels, is highly adaptable for different choices of complementary color emitting coordination complexes and Qdots.
- This article is part of the themed collection: Editors Collection for RSC Advances - India
 Please wait while we load your content...
Please wait while we load your content...

