
A thermal behavior and kinetics study of the catalytic pyrolysis of lignin
Abstract
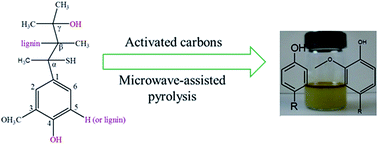
The aim of the present study is to convert lignin into bio-based phenols by catalytic pyrolysis using activated carbon (AC) as a catalyst. The thermal decomposition behavior of lignin pyrolysis was investigated using a thermogravimetric analyzer (TGA). The heating rate played a significant role in lignin thermal degradation, the mass loss of lignin in pyrolysis increased with the increase of heating rate. The reaction kinetics of lignin pyrolysis was determined and compared using microwave and conventional heating, a second-order reaction mechanism fitted well for lignin pyrolysis, and the results revealed that the activation energy for catalytic microwave pyrolysis of lignin was 7.32 kJ mol−1, which was remarkably lower than that for conventional pyrolysis of lignin (59.75 kJ mol−1). The reaction mechanism of this process was analyzed.
 Please wait while we load your content...
Please wait while we load your content...

