
Recent advances of titanium dioxide (TiO2) for green organic synthesis
Lee Eng Oia,
Min-Yee Chooa,
Hwei Voon Leea,
Hwai Chyuan Ongb,
Sharifah Bee Abd Hamida and
Joon Ching Juan*ac
aNanotechnology and Catalysis Research Center (NANOCAT), University of Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: jcjuan@um.edu.my; Fax: +60-3-7967-6556; Tel: +60-3-7967-6960
bDepartment of Mechanical Engineering, Faculty of Engineering, University of Malaya, 50603 Kuala Lumpur, Malaysia
cMonash University, Sunway Campus, Jalan Lagoon Selatan, 46150 Bandar Sunway, Selangor, Malaysia
First published on 15th November 2016
Abstract
Titanium dioxide (TiO2) has become increasingly popular as a catalyst. Although many applications of TiO2 involve photocatalysis and photoelectrochemical reactions, there are numerous interesting discoveries of TiO2 for other reactions. This review focuses on the recent development of TiO2 as a catalyst in green organic synthesis including in hydrodeoxygenation, hydrogenation, esterification/transesterification, the water–gas shift reaction, and visible light-induced organic transformation owing to its strong metal-support interaction (SMSI), high chemical stability, acidity, and high redox reaction at low temperature. The relationship between the catalytic performance and different metal or metal oxide dopants, and different polymorphs of TiO2 are discussed in detail. It is interesting to note that the reduction temperature and addition of promoters have a significant effect on the catalytic performance of TiO2.
1. Introduction
Titanium dioxide is known as titanium(IV) oxide or titania, with the chemical formula TiO2. Compared with bulk TiO2, TiO2 nanomaterials have been passionately studied over the past decades due to their potential applications in many areas. TiO2 is very useful because of its non-toxicity, chemical stability, low cost, and other advantages. TiO2 has been broadly used in paint as white pigments, cosmetics, food colouring, solar cells, photovoltaic cells, and photo applications. Many niche applications have also been explored especially in biomedical fields such as cancer therapies, orthopaedic surgery, stent placement, bactericides, and protein separation.1–3 Recently, research related to TiO2 catalysts in organic synthesis such as in deoxygenation, hydrogenation, the water–gas shift reaction, and CO oxidation has been carried out.4–7Pure TiO2 is either colourless or white, but it also tends to exist in other colours (yellow, red, brown, black, etc.) due to the presence of impurities (e.g. iron, niobium, chromium, tantalum, and vanadium). All three polymorphs of TiO2 have a very high refractive index and are temperately hard and dense. TiO2 is classified as non-hazardous according to the United Nations (UN); studies have revealed that ingestion of or skin or eye contact with TiO2 has no significant harmful effects to health.8 However, inhalation of TiO2 dust, especially at a nano-sized scale, can inflame the lungs. An occupational daily exposure limit of 15 mg of TiO2 dust has been set by the OSHA.8 Unlike other dust, TiO2 dust does not cause scarring as it is not chemically irritating to the lungs.
Various excellent reviews have been done on the synthesis methods of TiO2 and its application in photocatalysis and photoelectrochemistry; typical examples are on solar cells, hydrogen production by water splitting,9–11 degradation of organic pollutants,12–15 and reduction of carbon dioxide.16 However, there are limited reviews on the application of TiO2 as a support or catalyst for green organic synthesis. Thus, this paper reviews the challenges of TiO2 as a catalyst support and its application in various organic synthesis with a particular focus on: (1) deoxygenation; (2) hydrogenation; (3) esterification/transesterification; (4) the water–gas shift (WGS) reaction; and (5) visible light-induced organic transformation.
2. TiO2 crystal structure and properties
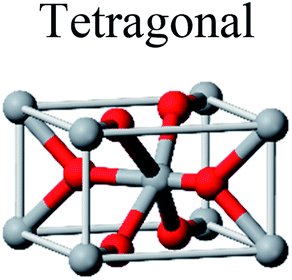
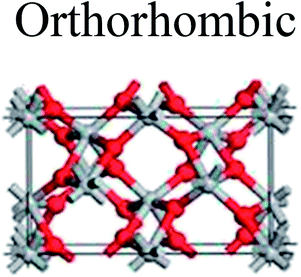
TiO2 has three main crystal polymorphs, namely anatase, rutile, and brookite. Each polymorph exhibits different physical properties. Among the three polymorphs, rutile is the most stable phase under ambient conditions while anatase and brookite are metastable at all temperatures and will transform to rutile when they are heated. Some research reported that the stability of various TiO2 phases was dependant on the particle size. Anatase is the most thermodynamically stable phase when the nanoparticles are smaller than 11 nm. Rutile has been found to be the most stable for nanoparticles greater than 35 nm in size while brookite is the most stable phase for nanoparticles between 11–35 nm in size.17Table 1 shows the properties of TiO2 in the anatase, rutile, and brookite phases. The structure of TiO2 is formed by chains of distorted TiO6 octahedra where each Ti atom is surrounded by 6 oxygen atoms. The three-dimensional stacking of the octahedra in anatase, rutile, and brookite is shown in Table 1. The unit cell of tetragonal anatase contains four TiO2 units (12 atoms), while the unit cell of tetragonal rutile contains two TiO2 units (6 atoms), and the unit cell of orthorhombic brookite contains eight TiO2 units (24 atoms). Thus, anatase has a smaller cell volume than that of rutile and brookite. Therefore, most practical work has been carried out with anatase or rutile. The existence of either or both of these phases leads to different activities for chemical reactions which is discussed later. Brookite is the least stable phase among the three due to an orthorhombic structure, thus it is difficult to synthesize.8 This also means that it is rarely used for most applications and is seldom studied.
| Property | Anatase | Rutile | Brookite | Ref. |
|---|---|---|---|---|
| Crystal bulk structure | 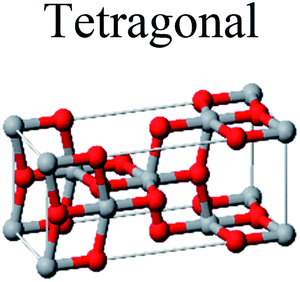 |
 |
 |
18 and 19 |
| Atoms per unit cell (Z) | 4 | 2 | 8 | 20 |
| Crystal size (nm) | <11 | >35 | 11–35 | 17 |
| Lattice parameters (nm) | a = b = 0.3785, c = 0.9514 | a = b = 0.4594, c = 0.2959 | a = c = 0.5436, b = 0.9166 | 19 and 21 |
| Space group | D194h – I4I/amd | D194h – P42/mnm | D152h – Pbca | 18 and 19 |
| Unit cell volume (nm3) | 0.1363 | 0.0624 | NA | 20 |
| Density (g cm−3) | 3.83 | 4.24 | 4.17 | 22 |
| Band gap (eV) | 3.26 | 3.05 | NA | 19 |
| Hardness (Mohs) | 5.5–6 | 6–6.5 | 5.5–6 | 22 |
The TiO2 phase is the most critical parameter determining the properties of the materials, therefore, many researchers attempted to understand the phase transformation between anatase, rutile, and brookite. The initial crystalline TiO2 phase formed is generally anatase; rutile is obtained through calcination.23 The transformation of the anatase to rutile phase is reconstructive and irreversible because the transformation involves bonds breaking and reforming.20 It was reported that the transformation of anatase to rutile occurs at approximately 600 °C when calcined in air.24–26 Nevertheless, some research reported that the transformation temperature may vary widely from 400 to 1200 °C with different raw materials, synthesis methods, and heat flow conditions.19,27–30 Brookite was transformed into the rutile phase when heated at about 800 °C. Rutile is the most thermally stable among the three polymorphs with a melting point between 1830 °C and 1850 °C.8 TiO2 is always difficult to dissolve or react when the sample is heated at above 1000 °C for a long period of time.8 Phase transformation is of considerable interest as the transformation is capable of altering the properties and performance of the catalyst.
3. TiO2 as catalyst support
Recently, the use of metallic nanoparticles for green and sustainable synthesis has attracted tremendous interest. In particular, the combination of metallic nanoparticles with a catalyst support is essential to enhance the catalytic activity and optimize selectivity in the desired application. Catalyst supports play an important role in improving the stability of the catalyst and reducing the amount of costly metal used.31 This is due to their unique properties, such as porosity, large surface area, tunable shapes, and high dispersity.32 Besides, a heterogeneous catalyst support has a significant impact on the catalytic activity and catalyst stability. It exerts a structural and active phase linked effect, which is able to increase the dispersion of metal species and inhibit the aggregation of active metal particles.33,34 Therefore, the selected heterogeneous catalyst support must retain its specific properties.According to the literature, catalyst supports can be grouped into four categories including (i) solid acid supports like zeolites,35 (ii) reducible oxide supports such as titania,36,37 ceria,38 and zirconia,39 (iii) refractory oxide supports like alumina33 and silica,38 and (iv) carbon-based supports like mesoporous carbon40 and carbon nanotubes.41 Different groups of support revealed different catalytic activity and selectivity towards the chemical reaction.
Among the reducible oxide supports, TiO2 is a famous and proven one for contributing to better catalytic activities as it acts as a promoter, a carrier for metals and metal oxides, and an additive or a catalyst.42,43 The better catalytic activity of TiO2 was strongly contributed to by the strong metal-support interaction (SMSI).44–46 SMSI was first observed by Tauster et al.47 in 1978; he defined the concept of SMSI as the drastic changes in the chemisorption properties of group 8–10 noble metals when supported on TiO2. Prototypical examples of SMSI phenomena were only observed for metallic particles supported on reducible oxides such as TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48,49 and CeO2.50 TiO2 exhibits SMSI after reduction at high temperature in the presence of a hydrogen environment. SMSI is an important and unique effect that occurs due to an electronic or geometrical interaction, and charge transfer between metallic nanoparticles and a support (Fig. 1).51,52 The metallic nanoparticles are capped with functional groups of the support that migrate to the surface of the metallic nanoparticles during the reduction process.50 This capping layer can form new active sites and improve the adsorptive and efficiency of the composite.53,54 The strength of the metal-support interaction can affect the metal particle dispersion on the support, the geometry of adsorption, and electronic structures, and significantly changes the catalytic performance.55,56 It also plays a dominant role in controlling the size growth of the metal.
48,49 and CeO2.50 TiO2 exhibits SMSI after reduction at high temperature in the presence of a hydrogen environment. SMSI is an important and unique effect that occurs due to an electronic or geometrical interaction, and charge transfer between metallic nanoparticles and a support (Fig. 1).51,52 The metallic nanoparticles are capped with functional groups of the support that migrate to the surface of the metallic nanoparticles during the reduction process.50 This capping layer can form new active sites and improve the adsorptive and efficiency of the composite.53,54 The strength of the metal-support interaction can affect the metal particle dispersion on the support, the geometry of adsorption, and electronic structures, and significantly changes the catalytic performance.55,56 It also plays a dominant role in controlling the size growth of the metal.
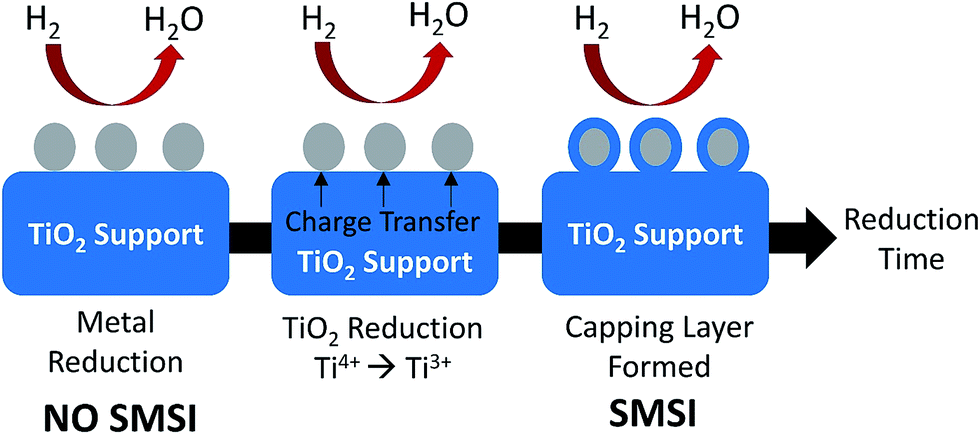 | ||
| Fig. 1 The SMSI effect occurring during reduction treatment by H2. The grey circles represent metallic nanoparticles, while the blue parts represent the TiO2 support. | ||
Besides the interaction of the metal and support, the size of the active sites is another factor that is pertinent to the efficiency of metal-supported catalysts. The size of the metal particle after heat treatment is highly dependent on the nature of the support. Fang et al. reported that gold particles with a mean cluster size of 1.5 nm were dispersed on TiO2, carbon, and SiO2 respectively. After calcination, the size of the gold particles increased significantly on carbon (4 nm) and SiO2 (5 nm); in sharp contrast, the gold particles on TiO2 (2 nm) were the smallest.57 This indicates that TiO2 effectively prevents the growth and sintering of metal particles.57 Thus, smaller metal particles can be obtained.
Moreover, the catalytic activity of metal-supported catalysts is attributed to two things: (1) contribution from the metal surface, and (2) other derives from the active sites, where the spill-over hydrogen could react with the adsorbed molecules.58 TiO2 acts as a reservoir of H atoms where H atoms may migrate from one reactant to another reactant. The hydrogen molecules adsorbed and dissociated on the metal surface then migrate to the TiO2 where they are trapped at surface defects. This migration will provide an active species for reaction. Then H+ on TiO2 can be exchanged with H2 easily in the presence of the metal phase.59 Fig. 2 demonstrates the route for hydrogen spill-over on acid sites.59 The hydrogen spill-over can greatly enhance reduction and the hydrogenation capability of TiO2. Therefore, TiO2 as a catalyst support not only acts as a platform for the dispersion of active species but also as an active site that impacts the catalytic performance.59
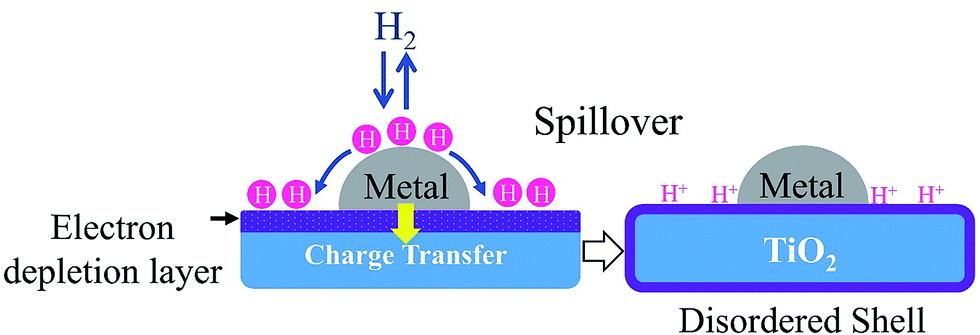 | ||
| Fig. 2 An example of hydrogen spill-over involved in hydrogenation of metal TiO2. | ||
TiO2 is well-known as a weak acid support, mainly with Lewis acid properties, in which Bronsted acidity is not observed.60 Lewis acid sites tend to attract the oxygen ion pair of the target molecules, therefore, this leads to higher selectivity for the hydrogenated product. Some studies showed that a weak acid support with Lewis acid sites from TiO2 was able to reduce coke formation or cracked products.58 It is well-known that coking on a catalyst surface caused by a polymerization reaction is one of the major challenges for the catalytic reaction, as coking significantly reduces the active sites by competition with the reactant. Therefore, catalyst deactivation can be minimized by using a TiO2-supported catalyst.
In addition, much research has shown that a variety of metal and non-metal dopants were capable of increasing the surface area and thermal stability of TiO2 through structural modifications and grain growth inhibition.44,61–64 It is important to select the appropriate synthetic routes to produce TiO2 with a higher surface area and more ideal porosity. Three-dimensional mesoporous TiO2 with a narrow pore size distribution and large surface area is able to increase the degree of dispersion and homogeneity of immobilized catalysts.65 Olsen et al.61 reported that mesoporous TiO2 synthesized by modification with aluminium was thermally stable up to 400 °C with a surface area of 479 m2 g−1. This is apparently the highest thermal stability with a large surface area. It is believed that modifying the TiO2 catalyst support definitely enhances the catalytic stability and activity of the catalyst.
4. Application of TiO2 catalyst for green organic synthesis
4.1 Deoxygenation
Deoxygenation involves the removal of oxygenated compounds from a molecule in the form of water, CO2, or CO. Deoxygenation can be accomplished with or without the presence of hydrogen during the reaction. Various possible mechanisms for deoxygenation (Table 2) include: (1) decarboxylation that removes carboxyl groups to yield paraffins and CO2; (2) decarbonylation that yields olefins, CO, and water; (3) hydrodeoxygenation that involves C–O bond cleavage under hydrogen and yields hydrocarbons and water, and for fatty acids, it involves reducing the oxidation state of the carbon atom of the carboxylic groups using H2; and (4) hydrogenolysis that involves cleavage of the carbon–carbon or carbon–heteroatom bond when hydrogen is added.66,67 High oxygenated compounds confer undesirable properties to bio-oil because they contribute to the high corrosiveness, low thermal stability, low volatility, and low energy content.68 Hence, oxygen removal is the most important process in the production of sustainable and high quality fuel components that are suitable to use with existing technology.| No. | Liquid phase | Precursor | End products |
|---|---|---|---|
| 1 | Decarboxylation | 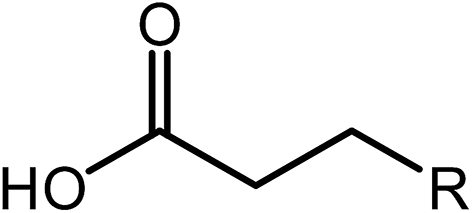 |
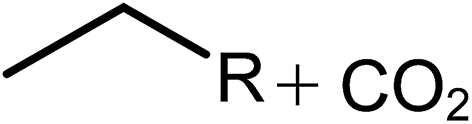 |
| 2 | Decarbonylation |  |
 |
| 3 | Hydrodeoxygenation | 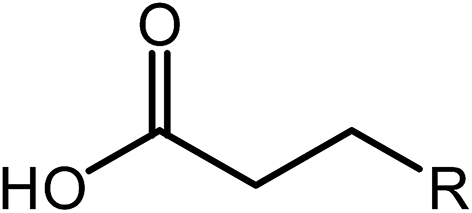 |
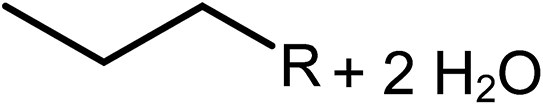 |
| 4 | Hydrogenolysis | 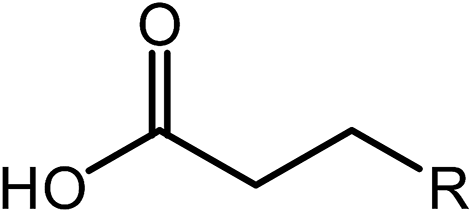 |
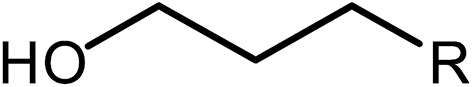 |
Table 3 shows an overview of the relevant literature for the deoxygenation of vegetable oil-based feedstocks and model compounds under different reaction conditions and the selectivity.
| No. | Catalyst | Metal (w/w) | Reactant | t (h) | T (°C) | Reaction conditions | BET (m2 g−1) | Phase | Conv. (%) | Selectivity | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Abbreviation: NA (not available in the reference); t (time); T (temperature); Conv. (conversion); PT (pre-treatment); P (pressure); A (anatase); R (rutile).b Phase was estimated using the calcination temperature. | |||||||||||
| 1 | TiO2 | NA | Guaiacol | 12 | 300 | PT = 15% H2S/H2/400 °C/4 h, PH2 = 4.0 MPa | 120 | Ab | 25 | 50% catechol, 35% phenol | 69 |
| 2 | CoMoS/TiO2 | 7.4% Mo | Guaiacol | 12 | 300 | PT = 15% H2S/H2/400 °C/4 h, PH2 = 4.0 MPa | 112 | Ab | 100 | 60% phenol, 20% benzene, cyclohexane & cyclohexene | 69 |
| 3 | NiMoS/TiO2 | 3.3% Ni, 15% Mo | Rapeseed oil | 4 | 300 | PH2 = 3.5 MPa | 117 | Ab | 100 | 56% C18, 5% C17 | 70 |
| 4 | Ru/TiO2 | 5% Ru | Pyrolysis oil | 4 | 350 | PH2 = 20 MPa | 55 | A:R |
100 | 67% oil | 71 |
| 5 | Ru/TiO2 | 1% Ru | Ethyl stearate | 17 | 200 | PH2 = 3.0 MPa, 5 mL hexane | NA | A | 99.3 | 91% C17, 8.9% C18, 0.1% stearyl alcohol | 72 |
| 6 | Ru/TiO2 | 4% Ru | Phenol | 1 | 300 | PT = N2/H2/400 °C/4 h, PH2 = 4.5 MPa | 33 | 3:1 A:R |
12 | 12% cyclohexanone, 2% cyclohexenol, 85% benzene, 1% cyclohexane, 0.6% cyclohexene | 52 |
| 7 | Pt/TiO2 | 1% Pt | m-Cresol | 1 | 300 | PT = H2/300 °C/1 h, PH2 = 0.1 MPa | NA | Ab | 17 | 88% toluene | 68 |
Deoxygenation based on TiO2 as a catalyst or catalyst support was usually pressurized with hydrogen. The presence of hydrogen was able to enhance the catalyst stability and avoid the formation of unsaturated hydrocarbon products.35,70 Accumulation of unsaturated hydrocarbon products and partially oxidized intermediates leading to coking occurs on the catalyst surface, thereby causing blockage to the active sites and hindering the catalyst.58,70 A weak acid support with Lewis acid sites such as TiO2 showed a low yield of coke or crack products.58 Coking is a major challenge for the deoxygenation reaction. It correlates with the structure of the catalyst as polymerization reactions are the major reactions leading to coking occurring on the surface of the catalyst.73 Therefore, this significantly eliminates the active sites. By using TiO2 as a catalyst or catalyst support, less coke is formed during the reaction, hence it is able to reduce catalyst deactivation.
TiO2 is a promising catalyst to catalyze the transformation of guaiacol through demethylation (Table 3, no. 1). The selectivity of the reaction was 50% catechol and 35% phenol, and hydrogenation was the main pathway observed. This contribution is attributed to the Lewis acid property of TiO2.74 When TiO2 was used as a support for the CoMoS catalyst, it has been clearly observed that CoMoS greatly enhanced the conversion rate and favoured selectivity to hydrodeoxygenation, in which phenol, benzene, and cyclohexane were the major products (Table 3, no. 2). The higher tendency towards hydrodeoxygenation is plausible due to a larger active phase cluster size that weakens the active phase support interaction.70 The hydrodeoxygenation reaction by TiO2-supported catalysts proceeds via two paths. Firstly, the phenol was directly deoxygenated to aromatic compounds as the primary product. Secondly, the ring was hydrogenated and then deoxygenated to a naphthenic product.75 It was believed that adsorption of oxygenated compounds onto TiO2 plays an important role in the transformation of phenolic compounds: a σ-bonding adsorption through the oxygen atom would give C–O hydrogenolysis while π-bonding of the aromatic ring would allow hydrogenation.75 The performance of TiO2 as a catalyst and catalyst support has been compared and it appeared that TiO2 as a catalyst support gave a very efficient conversion of guaiacol into deoxygenated hydrocarbons with a totally different selectivity. This demonstrated that the selectivity of TiO2 can be tuned by incorporation of a suitable active metal.
Several reports (Table 3, no. 2 and 3) showed that a metal sulphide catalyst such as nickel molybdenum (NiMo) and cobalt molybdenum (CoMo) supported on TiO2 showed high selectivity towards hydrodeoxygenation. Compared with sulphided CoMoS supported on Al2O3, the CoMoS did not facilitate the hydrodeoxygenation process and favoured direct cleavage of the aromatic carbon–heteroatom bond.74,76 This has confirmed that the selectivity of the catalyst could be fine-tuned by selection and modification of the support materials. For instance, Kubička et al.70 found that deoxygenation of rapeseed oil via NiMoS/TiO2 yielded n-octadecane (C18) as the main product and a minor share of n-heptadecane (C17) (Table 3, no. 3). The C18/C17 ratio for the deoxygenation of rapeseed oil achieved a value as high as ten. This has proven that the NiMoS/TiO2 catalyst exhibited high selectivity towards hydrodeoxygenation.
A metal sulphide catalyst supported on TiO2 in principle tackles two main reaction pathways, namely hydrodeoxygenation and hydrogenation. Generally, diesel fuel components are obtained selectively through hydrodeoxygenation via this catalyst, with the only by-products being water, carbon oxides, and propane.66 Due to the excellent performance, the catalysts are well-known and widely used in the industry. However, there are several disadvantages to this approach. The metal sulphide catalyst showed no activity for deoxygenation in hydrogen-free conditions, therefore, high hydrogen consumption is needed. In addition, an external sulfur source is needed to keep the sulfided catalyst in an active state and there is possible leaching of sulfur species in the final product. Hence, the most significant challenges from using metal sulphide catalysts, both from an environmental as well as an economical point of view, are the high hydrogen and sulfur consumption.
Ruthenium (Ru) is the common noble metal used in deoxygenation (Table 3, no. 4–6). For example, hydrodeoxygenation of pyrolysis oil with a Ru/TiO2 catalyst (Table 3, no. 4) achieved 100% conversion and 67% liquid product with a low oxygen content (about 9%).71 He et al.72 showed that the Ru/TiO2 catalyst successfully deoxygenated ethyl stearate to octadecane and heptadecane. Ethyl stearate was first hydrogenated to stearyl alcohol and then deoxygenated to octadecane and heptadecane. The most fascinating finding is that the selectivity towards heptadecane and octadecane is 91% and 8.9%, respectively. The total conversion of ethyl stearate is as high as 99.3% at 200 °C (Table 3, no. 5). On the other hand, Ru/TiO2 tended to follow the hydrogenolysis and hydrogenation pathway as shown in Table 3, no. 6. In the hydrogenolysis of phenol, the feedstock was reduced to benzene (about 85%). Meanwhile, the hydrogenation pathway firstly hydrogenated phenol to produce cyclohexenol. Cyclohexenol is an unstable product that rapidly tautomerizes to form cyclohexanone and is further converted to cyclohexanol in the presence of hydrogen. Cyclohexanol is then dehydrated to form cyclohexene and further hydrogenated to cyclohexane. The Ru/TiO2 catalyst shows outstanding activity and high selectivity toward direct hydrodeoxygenation. The superior performance can be explained by the synergistic action between small Ru particles and the TiO2 support. The role of TiO2 is explained by a spill-over mechanism in which small Ru particles facilitate hydrogen dissociation to create more active sites.77 The active sites are around the perimeter of the metal particles at the Ru/TiO2 interface, and also on the TiO2 support due to oxygen vacancies created from the spill-over mechanism.77 Spill-over hydrogen created in the periphery of Ru diffuses to the support in which Ti4+ was reduced to Ti3+ Lewis acid sites.72 Then Ti3+ interacted strongly with the carboxylic group or phenol hydroxyl group (Fig. 3),52 resulting in weakening the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and promoting hydrodeoxygenation. Therefore, it was believed that Ru/TiO2 could be a prominent catalyst in hydrodeoxygenation.
O bond and promoting hydrodeoxygenation. Therefore, it was believed that Ru/TiO2 could be a prominent catalyst in hydrodeoxygenation.
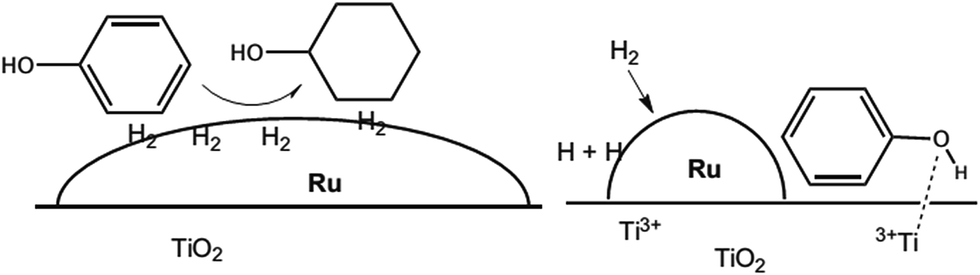 | ||
| Fig. 3 Illustration of large Ru particles on a TiO2 support interacting with the aromatic ring and facilitating hydrogenation (left), while small Ru particles create a bifunctional catalyst with the phenyl ring and promote hydrodeoxygenation (right) (adapted from ref. 49). | ||
Deoxygenation reactions in liquid phase based on various metals such as Ru, Pt, NiMo, and CoMo supported on TiO2 usually were studied at a temperatures ranging between 300 and 350 °C. Interestingly, the Ru/TiO2 catalyst showed a very high activity at a very low temperature of 200 °C, and the most attractive result is that the hydrocarbons C17 and C18 are produced via deoxygenation (Table 3, no. 5). These results are very striking in the upgrading of bio-oil due to both the low reaction temperature and high conversion rate and selectivity by using Ru/TiO2 as a catalyst.
In a separate study, Nie et al.68 reported that the selectivity of the catalyst to each of the possible pathways can be controlled by the selection of the metal and support. When using a noble metal such as platinum (Pt) supported on TiO2, hydrodeoxygenation becomes dominant. In the hydrodeoxygenation of m-cresol over Pt/TiO2, toluene is the main deoxygenation product (88%) (Table 3, no. 7). This can be explained by SMSI and stabilization of the carbonyl group at interfacial Pt–TiOx sites which are oxophilic. The oxophilic sites increase the rate of CO hydrogenation of m-cresol, and form an m-cresol tautomer (3-methyl-3,5-cyclohexadienone) as an intermediate which is further hydrogenated to a very reactive unsaturated alcohol (3-methyl-3,5-cyclohexadienol), and the alcohol is then dehydrated to toluene.68 It demonstrated that noble metal supports on TiO2 catalyst are promising for hydrodeoxygenation.
The TiO2 used for most of the hydrodeoxygenation studies was reported with a pure anatase crystalline structure. However, Wildschut et al.71 and Newman et al.52 reported that hydrodeoxygenation using P25 TiO2, a mixture of 75% anatase and 25% rutile, demonstrated good catalytic activity (Table 3, no. 4 and 6). This finding is in agreement with the result of Carballo et al.78 that Ru supported on P25 has a significantly better dispersion than that supported on pure anatase TiO2. The better metal dispersion is due to the rutile phase of P25 having a similar crystal structure to that of Ru.79 In addition, the rutile phase has a higher surface free energy than that of the anatase phase, and defect sites are likely to form on the surface of the rutile phase. The presence of defect sites on the rutile surface provides anchorage sites for the metal particles thereby inhibiting the metal particle size growth by coalescence or migration.77,80 Based on reduction kinetics of Ru supported on anatase and rutile, rutile was 2.5 times faster than anatase.79 According to Bickley et al., some cases have shown that small rutile crystallites are interwoven with the anatase crystallite and in some cases, the rutile phase forms a surface layer onto the anatase crystallites. The intimate contact of these two phases will enhance their interaction and further stabilize the metal particles.80 It was in accordance with the finding of Xaba and de Villiers, where the trend in sintering of metal on the phases of TiO2 is anatase > rutile > P25.81 This finding suggested that TiO2 with a combination of the anatase and rutile crystalline structures has better performance in hydrodeoxygenation.
4.2 Hydrogenation
Hydrogenation is the reaction where hydrogen addition occurs without the cleavage of bonds, as depicted in Fig. 4. The process is commonly used to reduce or saturate organic compounds under a hydrogen atmosphere. It is an essential process in the biofuel industry as biomass-derived compounds need to be hydrogenated or hydrodeoxygenated to produce biofuels and chemicals. Metal supported on TiO2 showed a high selectivity for hydrogenated products due to the Lewis acid properties.60 In addition, they have a very low yield of coked or cracked products.58 Hence, more studies have been focused on TiO2-supported catalysts in hydrogenation.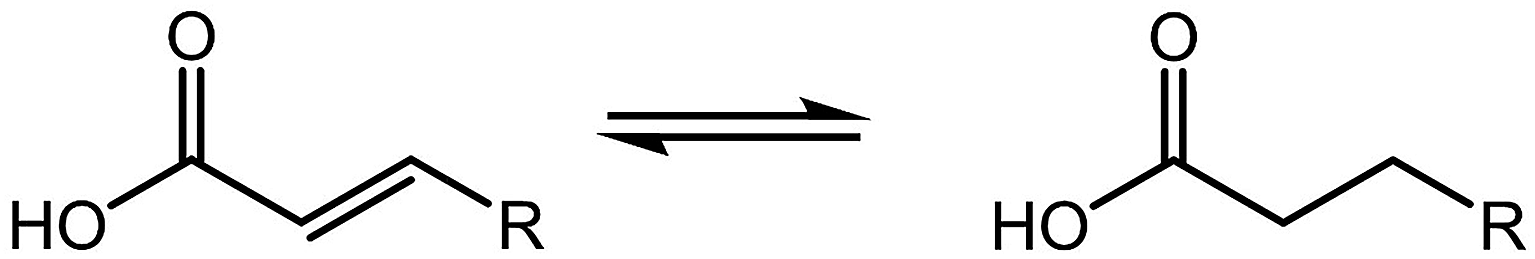 | ||
| Fig. 4 Hydrogenation reaction of a carboxylic acid. | ||
Table 4 shows an overview of the relevant literature for the hydrogenation of various model compounds under a hydrogen atmosphere. It was shown that anatase, rutile, and a mixed phase of TiO2 have revealed good activity when combined with different metals and then reacted with different feedstocks.
| No. | Catalyst | Metal (w/w) | Reactant | t (h) | T (°C) | Reaction conditions | Phase | Conv. (%) | Selectivity | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Abbreviation: NA (not available in the reference); t (time); T (temperature); Conv. (conversion); PT (pre-treatment); P (pressure); A (anatase); R (rutile). | ||||||||||
| 1 | Pd/TiO2 | 1% Pd | Acetylene in ethylene | 1 | 40 | PT = H2, 300 °C/1 h, PH2 = 0.1 MPa | 3:1 A:R |
54 | 1% ethylene | 82 |
| 2 | Pd/TiO2 | 1% Pd | Acetylene in ethylene | 1 | 40 | PT = H2, 500 °C/1 h, PH2 = 0.1 MPa | 3:1 A:R |
56 | 25% ethylene | 82 |
| 3 | Pd/TiO2 | 0.075% Pd | Alkadienes (C10–C13) | 1 | 100 | PT = H2, 200 °C/1 h, PH2 = 1.4 MPa | A | 72.6 | 72.4% alkenes | 83 |
| 4 | Pd/TiO2 | 0.075% Pd | Alkadienes (C10–C13) | 1 | 100 | PT = H2, 200 °C/1 h, PH2 = 1.4 MPa | R | 62.6 | 34.7% alkenes | 83 |
| 5 | Pd/TiO2 | 0.075% Pd | Alkadienes (C10–C13) | 1 | 100 | PT = H2, 450 °C/1 h, PH2 = 1.4 MPa | R | 69.5 | 86.4% alkenes | 83 |
| 6 | Pt/TiO2 | 4% Pt | Stearic acid | 20 | 130 | PT = H2, 120 °C/1 h, PH2 = 2.0 MPa | A | 100 | 93% stearyl alcohol, 7% Cn + Cn−1 | 42 |
| 7 | PtRe/TiO2 | 4% Pt, 4% Re | Stearic acid | 4 | 130 | PT = H2, 120 °C/1 h, PH2 = 2.0 MPa | A | 100 | 70% stearyl alcohol, 30% Cn + Cn−1 | 42 |
| 8 | PtIr/TiO2 | 0.5% Pt, 1% Ir | Cyclohexene | 1 | 100 | PT = H2/N2, 400 °C/1 h, PH2 = 0.1 MPa | A | 100 | 100% cyclohexane | 84 |
| 9 | Ni/TiO2 | 10% Ni | Benzaldehyde | 6 | 140 | PT = H2, 350 °C/16 h, PH2 = 0.1 MPa | R | 100 | 5% benzyl alcohol, 64% toluene, 31% benzene | 38 |
| 10 | Ni/TiO2 | 15% Ni | Acetophenone | 1 | 140 | PT = H2, 450 °C/4 h, PH2 = 3.9 MPa | R | 96.7 | 100% phenylethanol | 85 |
| 11 | Ni/TiO2 | 8% Ni | Ethanol | 8 | 500 | PT = H2/N2, 500 °C/1 h, PN2/He = 0.1 MPa | A | 80 | 0.21 mol min−1 of H2, 19% CH3CHO, 6% CH4 | 86 |
| 12 | Ni/TiO2 | 8% Ni | Ethanol | 8 | 500 | PT = H2/N2, 800 °C/1 h, PN2/He = 0.1 MPa | R | 86 | 0.44 mol min−1 of H2, 5.9% CH3CHO, 13.1% CH4 | 86 |
The catalytic activity of different metals supported on TiO2 yielded different hydrogenation efficiency and product selectivity. As shown in Table 4, noble metals such as palladium (Pd), platinum (Pt), and iridium (Ir) have attracted much attention due to the difference in the catalytic activity and selectivity of hydrogenation. For instance, Pd/TiO2 showed a high activity (72.4%) for alkene production from alkadienes via hydrogenation (Table 4, no. 3), while other reports showed that Pt/TiO2 also achieved outstanding catalytic activity in hydrogenation reactions, which is 100% conversion (Table 4, no. 6–8). Manyar et al.42 revealed a facile hydrogenation of carboxylic acids using Pt/TiO2, with 100% conversion and high alcohol selectivity of 93% and 7% of linear alkanes under a low temperature and hydrogen pressure. The enhanced hydrogenation activity could be explained by the SMSI that occurs between noble metals and reducible oxides such as TiO2. The oxygen vacancies on TiO2 that are created through the hydrogen spill-over interact with the carbonyl oxygen of the acid (Fig. 5). The interaction weakens CO and promotes hydrogenation and CO bond cleavage.42 Therefore, this finding confirmed that SMSI between noble metals and TiO2 is the key factor affecting the hydrogenation activity and catalyst selectivity.
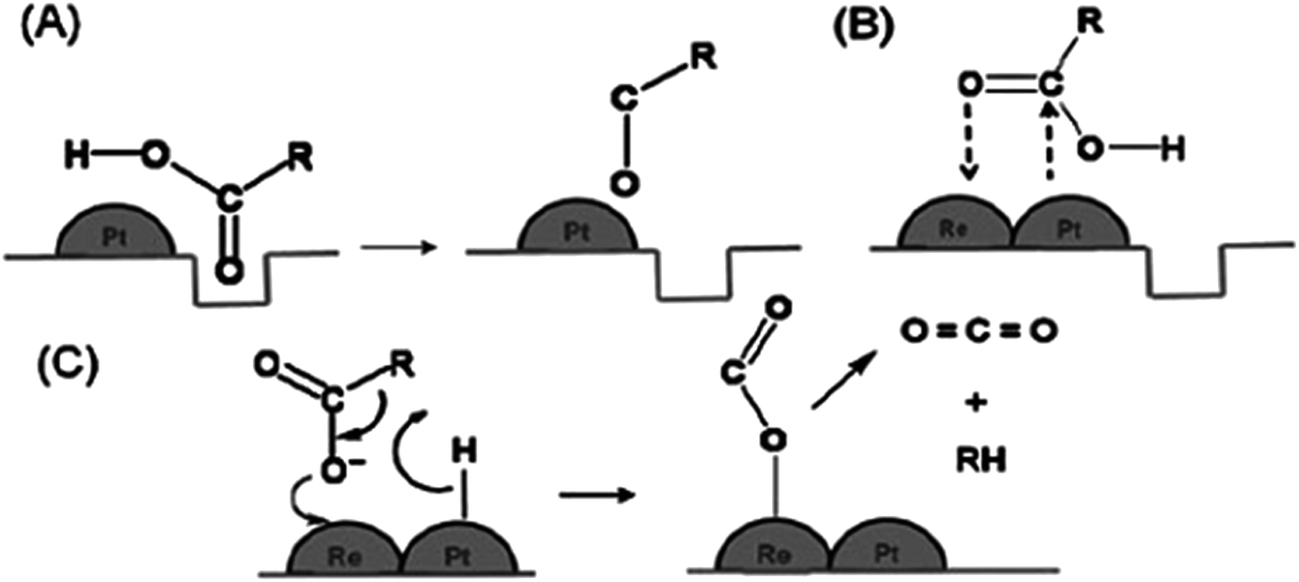 | ||
| Fig. 5 Hydrogenation of carboxylic acid ensuing through (A) the interaction of the oxygen atom of the carbonyl group with the oxygen vacancies created by hydrogen spill-over by Pt metal, (B) the interaction of the oxygen atom of the carbonyl group with Re on the surface of the PtRe/TiO2 bimetallic catalyst, and (C) the decarboxylation of carboxylic acid on the surface of Re on the surface of the PtRe/TiO2 bimetallic catalyst (adapted from ref. 40). | ||
Nickel (Ni) is another metal catalyst for hydrogenation study (Table 4, no. 9–12) due to its cost effectiveness compared to noble metals and ease of separation from homogeneous media by a magnetic field.87 Saadi et al.38 demonstrated the highest conversion and selectivity for benzaldehyde to 64% toluene and 31% benzene (Table 4, no. 9). This suggested that Ni/TiO2 preferentially hydrogenates the CO double bond to form benzyl alcohol and toluene (about 69% for both hydrogenated products). While the C–C exocyclic bond of benzaldehyde undergoes hydrogenolysis to form benzene. This observed selectivity was explained by the existence of SMSI between Ni and TiO2, and the existence of SMSI promotes dispersion of Ni.88 Moreover, strong reducibility and surface acid–base properties of TiO2 increase the hydrogenation selectivity.38,43 Thus, the Ni/TiO2 catalyst is a potential non-noble metal catalyst for hydrogenation and hydrogenolysis.
As compared to the hydrogenation with monometallic Pt/TiO2 to that with bimetallic PtRe/TiO2, the addition of rhenium (Re) increased the rate of hydrogenation and thus significantly increased the selectivity towards linear alkanes by 23%. Surprisingly, the reaction duration with PtRe/TiO2 to completion was 5 times faster than that with monometallic Pt/TiO2. The presence of Re increased oxyphilicity because the Re interacts with the lone pair electrons of the carbonyl group oxygen (Table 4, no. 7). The interaction of the substrate with Re on the catalyst surface favours the formation of alkanes.42 Other studies reported that bimetallic PtIr/TiO2 showed a higher hydrogenation ability than Pt/TiO2 did. It can be explained by the high dispersion of Pt or Ir on TiO2 and the mean size of PtIr is 2.1 nm smaller than Pt alone. It was found that Ir particles strongly interacted with TiO2 and acted as nucleation sites for Pt atom assembly.84 Therefore, the bimetallic catalyst could be an alternative to a monometallic catalyst for more active hydrogenation and can shorten the reaction duration.
Interestingly, the reduction temperature of the catalyst also plays an important role in stabilizing the catalyst to achieve a higher hydrogenation rate. As discussed above, the hydrogenation reaction on the TiO2-supported catalyst depends on the strong metal-support interaction (SMSI). It was believed that the reduction temperature affects the SMSI of TiO2-supported catalysts. This was confirmed by Kang et al.82 where Pd interacted strongly with TiO2 particularly after reduction at high temperatures due to SMSI. By comparing Pd/TiO2 reduced at 300 °C and 500 °C (Table 4, no. 1 and 2), it was found that Pd/TiO2 reduced at 500 °C induced SMSI and revealed higher conversion and selectivity of hydrogenation from acetylene to ethylene. Li et al.83 also reported that SMSI corresponding to noble metals supported on TiO2 was only observed when the catalyst was reduced at a temperature above 300 °C. In addition, this finding was further proven by Wildschut et al.71 where SMSI exists in between Pd and anatase TiO2 at a low reduction temperature (Table 4, no. 3). However, SMSI for Pd supported on rutile TiO2 was only observed when the catalyst was reduced at a higher temperature because rutile is thermodynamically and structurally more stable than anatase. Pd/rutile TiO2 reduced at 200 °C does not induce SMSI and hence the conversion and selectivity are low. The elevation of the reduction temperature from 200 °C to 450 °C leads to the presence of SMSI between Pd and Ti3+ in rutile TiO2. The presence of SMSI has enhanced the conversion of alkadienes and selectivity of hydrogenation by 52%. Therefore, it is important to reduce the catalyst at high temperature prior to reaction to induce the SMSI phenomenon for improving selectivity.
Furthermore, some research showed that TiO2 in different polymorphs exhibits different physicochemical properties which influenced the catalytic activities. Hydrogenation of acetophenone with Ni/rutile TiO2 exhibits prominent activity (96.7% conversion and 100% selectivity) as compared to that of Ni/anatase TiO2 (Table 4, no. 10). In separate studies, Nichele et al.86 highlighted that Ni/rutile TiO2 demonstrated high ethanol conversion (86%) as compared to that of Ni/anatase TiO2 (80%). The higher activity of Ni/rutile TiO2 is essentially attributed to SMSI between Ni and rutile, arising from the higher surface enthalpy of rutile than that of anatase.85 This has prevented the aggregation of Ni particles leading to the higher dispersion of Ni on the surface of rutile TiO2 which led to a higher activity.85,88,89 Besides, electron density on Ni is altered due to electron transfer between Ni and rutile. The interaction between Ni and Ti3+ may generate a negative charge on Ni atoms that enhances the activity of the Ni/rutile TiO2 catalyst for the hydrogenation.52 Thereby, it is affirmed that rutile TiO2 is more active as compared to anatase TiO2 in hydrogenation.
In addition, the hydrogenation activity also depends on the morphology of the catalyst support and particle size distribution.84 Bimetallic PtIr/TiO2 nanotubes (Table 4, no. 8) exhibited excellent hydrogenation of cyclohexene to cyclohexane with 100% conversion and the unique properties of TiO2 nanotubes have enhanced the hydrogenation process. For example, the Pd/TiO2 nanotubes and Pt/TiO2 nanotubes showed a better response and high selectivity towards double bond hydrogenation.51,90 TiO2 nanotubes are regarded as an outstanding catalyst support because they exhibit a large internal and external surface, along with available surface in the vertex and in the interlayer. In line with this, the nanotubes have a bigger specific surface area (SSA) for the high population of exposed Ti–OH. The SSA of TiO2 nanotubes is almost twice the size of TiO2 nanoparticles.90,91 This is also suggested by the fact that the surface area and the number of adsorption sites have increased extensively. Hence, it is believed that TiO2 nanotubes have better adsorption and performance in hydrogenation.
4.3 Esterification and transesterification
Esterification is a reaction between alcohol with acid to produce ester and water, while transesterification is the reaction of an ester with alcohol to replace the alkoxy group (Fig. 6). Catalysts with basicity perform better in transesterification than an acid catalyst does. Transesterification of triglycerides with short chain alcohol is the most common method to produce biodiesel. It is relatively simple and has easy operation, but the product as the transportation fuel suffers from engine compatibility issues due to the incomplete removal of oxygenates.92 Meanwhile, esterification is a chemical process to convert platform molecules into renewable base chemicals (alkyl ester) in the presence of an acid catalyst.93,94 Both esterification and transesterification are an important technology in the chemical and fuel industries.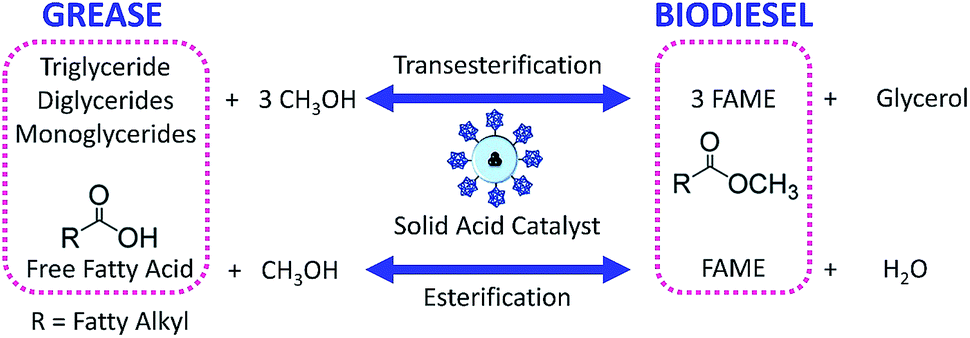 | ||
| Fig. 6 Esterification of fatty acids and transesterification of triglyceride to produce biodiesel. | ||
Acid-modified metal oxides such as sulfated TiO2 (TiO2–SO4), sulfated zirconium dioxide (ZrO2–SO4), and sulfated iron oxide (Fe2O3–SO4) have been studied as potential catalysts for the last two decades in different types of reactions. Lin et al.95 reported that TiO2–SO4 catalyzed the esterification reaction between acetic acid and cyclohexanol well (Table 5, no. 1), whereas TiO2 alone was not reactive towards the esterification reaction. In another study, it was shown that the esterification of levulinic acid to ethyl levulinate using TiO2–SO4 achieved as high as 83.2% conversion (Table 5, no. 2). Besides, transesterification of soybean oil and castor oil using TiO2–SO4 was also reported; the catalyst exhibited high transesterification conversion of 40% and 25%, respectively (Table 5, no. 4 and 5). Sulfate on TiO2 plays an important role in esterification/transesterification by offering active acid sites. The sulfate linked to TiO2 on the chelate form and induced Bronsted acid sites (Fig. 7). The existence of strong Bronsted acid sites on TiO2–SO4 promotes the esterification/transesterification reaction.97,99 This clearly showed that the acid sites comprised of Lewis and Bronsted sites are crucial for the esterification/transesterification reaction.
| No. | Catalyst | Metal (w/w) | Reactant | t (h) | T (°C) | Reaction conditions | TiO2 morphology | Phase | Conversion/selectivity | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Abbreviation: NA (not available in the reference); t (time); T (temperature); Conv. (conversion); P (pressure); A (anatase); R (rutile).b Estimated value based on mol ratio. | ||||||||||
| 1 | TiO2–SO4 | NA | Acetic acid | 10 | 100 | NA | Nanotubes | A | 55% | 95 |
| 2 | TiO2–SO4 | NA | Levulinic acid | 3 | 105 | NA | Nanorods | A | 83.2% ethyl levulinate | 96 |
| 3 | ZrO2/TiO2–SO4 | 20% ZrO2 | Levulinic acid | 3 | 105 | NA | Nanorods | A | 90.4% ethyl levulinate | 96 |
| 4 | TiO2–SO4 | 17% H2SO4b | Soybean oil | 1 | 120 | P = 0.5 MPa | Mesoporous | A | 40% FAMEs | 97 |
| 5 | TiO2–SO4 | 17% H2SO4b | Castor oil | 1 | 120 | P = 0.5 MPa | Mesoporous | A | 25% FAMEs | 97 |
| 6 | Au/TiO2 | 1% Au | 5-hydromethylfurfural (HMF) + methanol | 3 | 130 | P = 0.3 MPa | NA | NA | 98% 2,5-furandimethylcarboxylate | 98 |
| 7 | Au/TiO2 | 1% Au | 5-hydromethylfurfural (HMF) + methanol | 3 | r.t. | P = 0.3 MPa | NA | NA | No conversion | 98 |
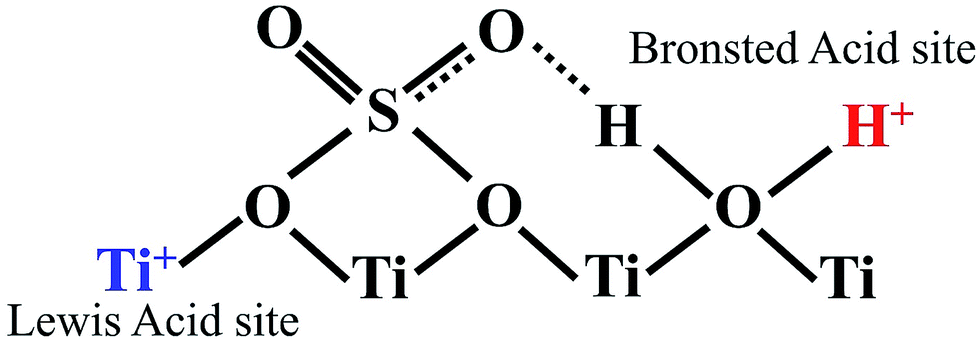 | ||
| Fig. 7 Illustrative structure indicates the presence of both Bronsted and Lewis acid sites on the surface of TiO2–SO4. | ||
Additionally, modification of a TiO2 support with a metal oxide such as ZrO2 is capable of enhancing the esterification reaction. TiO2–SO4 and zirconia-modified TiO2–SO4 (ZrO2/TiO2–SO4) was used for esterification of levulinic acid (Table 5, no. 2 and 3). It was observed that ZrO2/TiO2–SO4 enhanced the esterification of levulinic acid to ethyl levulinate by 30.4% as compared to that of unmodified TiO2. The reaction reached equilibrium for ZrO2/TiO2–SO4 with a levulinic acid conversion of 90.4% in 3.5 hours, while in the case of the reaction catalyzed by TiO2–SO4, the conversion is 7.2% lower than with ZrO2/TiO2–SO4.96 The author claimed that ZrO2 was highly dispersed on the surface of TiO2 using powder X-ray diffraction (XRD), scanning electron microscopy (SEM), and transmission electron microscopy (TEM). It was assumed that introduction of ZrO2 to TiO2–SO4 increased the adsorption of sulfate on the surface of TiO2 and acted as active sites for esterification.96 This clearly showed that TiO2 with modification exhibited an increased reaction for esterification and achieved equilibrium in a shorter reaction time.
Gold (Au) is another excellent catalyst for oxidative esterification. For instance, Taarning et al.98 demonstrated esterification of a primary alcohol in hydromethylfurfural (HMF) with Au/TiO2 at 130 °C for 3 hours. Furan-2,5-dimethylcarboxylate (FDMC) is formed directly with an excellent yield of approximately 98% (Table 5, no. 6). Besides, oxidation of HMF with Au/TiO2 at room temperature (22 °C) under oxygen atmosphere produced 90% of 5-hydroxymethylfuroate (HMMF) within 5 hours. Highly dispersed Au on a TiO2 support has been shown to be efficient for esterification and oxidation of HMF with remarkable selectivity.98 Thereby, Au/TiO2 offers an alternative to other oxidation catalysts for esterification and transesterification.
The morphological properties of a catalyst such as SSA, the mean pore size, and pore distribution are crucial parameters that influence the conversion and selectivity of esterification and transesterification. The specific morphology of the catalysts included nanorods, nanotubes, mesoporous materials, nanoparticles, etc. Several studies discussed the outstanding performance of nanotubes in esterification and transesterification (Table 5, no. 1–3). Nanotubes have larger internal and external surfaces, which reveal better adsorption sites. Moreover, mesoporous TiO2 with a bigger SSA and mean pore size featured transesterification with a higher yield (Table 5, no. 4 and 5). Therefore, it is of utmost importance to select a synthesis method with a specific morphology to obtain optimal catalytic performance.
4.4 Water–gas shift reaction
The water–gas shift (WGS) reaction is the main reaction used for the production of hydrogen by converting carbon monoxide in the presence of water. It is always used in conjunction with the steam reformation of methane or other hydrocarbons to produce hydrogen gas.100,101 The WGS reaction is also one of the important reactions used in the Fischer–Tropsch and methanation reaction to balance the hydrogen to carbon monoxide ratio. Recently, the interest for the WGS reaction as an integral component of fuel processing for hydrogen in fuel cell applications has grown significantly. With the high demand for clean fuel and hydrogen fuel cells, the development of WGS catalysts is one of the current research interests.(1) Steam reforming
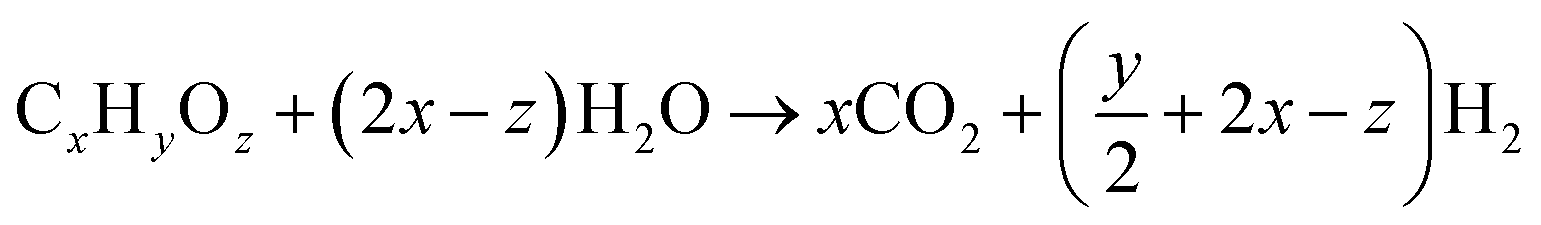
(2) Water–gas shift reaction
| CO + H2O ↔ CO2 + H2 |
(3) Fischer–Tropsch reaction
| (2n + 1)H2 + nCO → CnH(2n+2) + nH2O |
(4) Methanation reaction
| CO + 3H2 ↔ CH4 + H2O |
The WGS reaction normally involves both an active metallic phase and support. A redox process is one of the mechanisms proposed, whereby CO adsorbed on the metal and is oxidized by the catalyst support.101 The other WGS mechanism involves formates or carbonates on the catalyst surface as intermediates that decompose into hydrogen and carbonate, prior to CO2 liberation. Formates or carbonates are produced from the CO reaction with active hydroxyl from supports.34,105 Hence, it is clear that the catalyst support plays an important role in determining the WGS reaction owing to its role in dispersing metal on the support and the potential to reversibly exchange oxygen ions during the redox reaction.
The redox process of the WGS reaction primarily depends on the surface properties of the TiO2 support. Noble metals supported on TiO2 have been reported as promising low temperature WGS catalysts due to a high redox capability, resulting in the creation of oxygen vacancies (defects) through reduction of the cation charge (Tix+, x < 4) with CO.34,106 The oxygen vacancies could dissociate H2O to H2, regenerate surface oxygen sites, and form hydroxyl groups.107 As mentioned earlier, the surface properties of TiO2 are modifiable by combination with other transition metals or rare earth oxides. Panagiotopoulou et al.103 found that the conversion of CO at low temperature (<300 °C) was notably improved when Pt was dispersed on TiO2. The turnover frequency of CO increases exponentially when the primary crystallite size of TiO2 decreases, and this is accompanied by a considerable reduction of the apparent activation energy of the reaction.103 The noble metal catalysts highly dispersed on TiO2 are promising candidates for use in low temperature WGS reactors for fuel cell applications.
A noble metal such as platinum (Pt), rhodium (Rh), ruthenium (Ru), and palladium (Pd) supported on TiO2 is very effective for the WGS reaction (Table 6, no. 1–10). Among the various noble metals supported on TiO2, the CO conversion and turnover frequency (TOF) for the WGS reaction follow the order Pt > Rh > Ru > Pd (Table 6, no. 2–5). Pt is about 20 times more active than Pd. Pt/TiO2 was one of the most studied WGS catalysts due to the outstanding performance in hydrogen selectivity (Table 6, no. 5–10) and high redox properties.34,106 Pt/TiO2 is able to pass through both the classical redox route and the associative formate route for the WGS reaction. Thereby, Pt/TiO2 is an auspicious catalyst support for the WGS reaction at low temperature.
| No. | Catalyst | Metal (w/w) | t (h) | T (°C) | Reaction conditions | BET (m2 g−1) | Phase | Conv. (%) | TOF (s−1) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Abbreviation: NA (not available); t (time); T (temperature); Conv. (conversion); TOF (turnover frequency); PT (pre-treatment); P (pressure); A (anatase); R (rutile). | ||||||||||
| 1 | Ru/TiO2 | 2% Ru | 0.03 | 700 | 24 MPa | 49 | NA | 82 | NA | 102 |
| 2 | Pd/TiO2 | 0.5% Pd | 1 | 250 | PT = 300 °C/2 h/H2, PH2 = 0.1 MPa, 200 cm3 min−1 | 39 | 3:1 A:R |
2 | 0.027 | 103 |
| 3 | Ru/TiO2 | 0.5% Ru | 1 | 250 | PT = 300 °C/2 h/H2, PH2 = 0.1 MPa, 200 cm3 min−1 | 39 | 3:1 A:R |
6 | 0.07 | 103 |
| 4 | Rh/TiO2 | 0.5% Rh | 1 | 250 | PT = 300 °C/2 h/H2, PH2 = 0.1 MPa, 200 cm3 min−1 | 39 | 3:1 A:R |
8 | 0.16 | 103 |
| 5 | Pt/TiO2 | 0.5% Pt | 1 | 250 | PT = 300 °C/2 h/H2, PH2 = 0.1 MPa, 200 cm3 min−1 | 39 | 3:1 A:R |
22 | 0.46 | 103 |
| 6 | Pt/TiO2 | 0.5% Pt | 1 | 250 | PT = 300 °C/2 h/H2, PH2 = 0.1 MPa, 200 cm3 min−1 | 71 | A | 34 | 7 | 103 |
| 7 | Pt/TiO2 | 3% Pt | 1 | 250 | PT = 500 °C/2 h/H2, PH2 = 0.1 MPa, 9500 L h−1 kgcat | 74 | A | 33 | 82.4 × 102 | 104 |
| 8 | Pt/TiO2 | 3% Pt | 1 | 250 | PT = 500 °C/2 h/H2, PH2 = 0.1 MPa, 9500 L h−1 kgcat | 44 | R | 52.3 | 84.9 × 102 | 104 |
| 9 | Pt/TiO2 | 0.5% Pt | 1 | 300 | PT = 300 °C/1 h/H2/He, PH2 = 0.1 MPa, 41000 L h−1 kgcat |
NA | 3:1 A:R |
60 | 10 | 100 |
| 10 | PtCe/TiO2 | 6.6% Ce, 0.5% Pt | 1 | 300 | PT = 275 °C/2 h/H2/N2, PH2 = 0.1 MPa, 21200 L h−1 kgcat |
62 | NA | 90 | NA | 105 |
As discussed in other chemical reactions (Sections 4.1 to 4.3), TiO2 in different polymorphs has a different effect on the catalytic activity, and the WGS reaction is no exception. Anatase and Degussa P25 (a mixture of 75% anatase and 25% rutile) are common TiO2 polymorphs used for the study of the WGS reaction. Iida et al.104 observed that the Pt/rutile TiO2 catalyst (Table 6, no. 8) has a relatively high activity for the WGS reaction at low temperature. Pt/rutile TiO2 achieved 52.3% conversion which is 19.3% higher than that for Pt/anatase TiO2 (Table 6, no. 7). The superior catalytic activity was mainly attributed to the SMSI between Pt and TiO2. Besides, it was also mentioned that Pt/rutile TiO2 had a larger dispersion than Pt/anatase TiO2. A linear relationship was shown between the catalytic activity, SMSI, and Pt dispersion. This is in accordance with the previous report which stated that rutile revealed a greater catalytic activity than anatase due to better dispersion on rutile.78,104 The better dispersion can be explained by a high degree of lattice matching, ensuring better interaction between Pt and TiO2, preventing the agglomeration of Pt particles during the calcination.108 This shows that the efficiency of the catalyst also depends significantly on the crystal structure of the support.
The Pt/TiO2 catalysts are promising candidates for the WGS reaction. A report showed cerium (Ce)-modified TiO2 as a new support of Pt for the WGS reaction. Ce is a particularly interesting substance to be mixed with TiO2 substrates because Ce ions have useful surface properties for the WGS reaction. The addition of Ce to Pt/TiO2 increased the hydroxyl concentration on the TiO2 surface, and consequently induced a higher activity (approximately 90%) at the low temperature WGS reaction (Table 6, no. 10). The addition of Ce affects the redox properties of TiO2 because Ce was well dispersed and interacted with Ti atoms. Besides, Ti4+ ions can be replaced by Ce4+ ions to improve the thermal stability and redox properties of TiO2.105 The bimetallic PtCe/TiO2 was found to be highly active and stable in the WGS reaction, which indicated that it is a promising formulation as a WGS catalyst.
4.5 Visible light-induced organic transformation
Heterogeneous visible light photocatalysts have gained much attention recently in organic synthesis due to their simple chemical work-up and easy recovery. Solar energy is comprised of three main components, i.e. ultraviolet (λ = 200–400 nm), visible light (λ = 400–800 nm), and infrared (λ > 800 nm).109 Visible light is abundant in nature compared to ultraviolet and infrared, however it cannot be adsorbed directly by the reactant molecules to drive the reaction.110 Hence, visible light photocatalysts are needed as a bridging media for the energy transfer between the substrates and visible light. In the past decades, TiO2, a semiconductor with a relatively large band gap (3.2 eV), has been known to be an excellent photocatalyst under irradiation of ultraviolet light.111,112 The most recent research found that TiO2 also performed well as a photocatalyst under visible light irradiation.113,114Higashimoto et al. first reported the selective photocatalytic oxidation of benzyl alcohol to aldehydes under visible light irradiation; the conversion rate and selectivity to aldehydes was as high as 99% on TiO2 in the presence of O2 (Table 7, no. 1).115 Modified TiO2 with rhodium ions (Rh3+) also catalyzes the oxidation of benzyl alcohol in the presence of visible light and O2 (Table 7, no. 2). Benzaldehyde was obtained in a high yield (97%) with 100% conversion of benzyl alcohol. The visible light response is derived from the surface complexes formed when the alcohol was absorbed by the hydroxyl groups on the surface of TiO2.116 This research indicated the direct correlation between benzaldehyde formation and specific surface area, and the conversion rate increased with an increase in the surface area, showing that the surface area of the TiO2 supports was a decisive factor controlling the conversion rate.116
| No. | Catalyst | Reactant | t (h) | T (°C) | Reaction conditions | Oxidant | Phase | BET | Conv. (%) | Selectivity | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Abbreviation: NA (not available); t (time); T (temperature); r.t. (room temperature); Conv. (conversion); A (anatase); R (rutile). | |||||||||||
| 1 | TiO2 | Benzyl alcohol | 4 | r.t. | Visible light | O2 | A | NA | 99 | 99% benzaldehyde | 115 |
| 2 | Rh3+/TiO2 | Benzyl alcohol | 30 | r.t. | Visible light | O2 | A:R |
98 | 100 | 97% benzaldehyde | 116 |
| 3 | TiO2 | Benzylic amines | 4 | r.t. | Visible light | 1 atm air | A | 135 | 54 | 98% imine | 113 |
| 4 | TiO2 | Benzylic amines | 4 | r.t. | Visible light | 2 atm O2 | A | 135 | 76 | 98% imine | 113 |
| 5 | TiO2 | Benzylic amines | 4 | r.t. | Visible light | 1 atm air | A:R |
54 | 35 | 98% imine | 113 |
Oxidation of amines by TiO2 under visible light irradiation showed that with 1 atm of air as oxidant, the reaction proceeded smoothly with a conversion rate of 54% to produce the imine with 98% selectivity (Table 7, no. 3). When 2 atm of pure dioxygen was used as the oxidant, the conversion rate increased to 76% without having any influence on the selectivity (Table 7, no. 4). Hence, the purity of the oxidant affects the conversion rate of the photo-oxidation reaction. Nevertheless, atmospheric dioxygen is the most abundant oxidant, and a reaction carried out under air is greener. In addition, the surface area is also one of the important factors that affects the photocatalytic activity. Degussa P25 TiO2 (85% anatase and 15% rutile) usually has the highest activity for the reaction under ultraviolet irradiation.112,113 However, in the reaction under visible light irradiation, it is interesting to find out that Degussa P25 has a lower conversion rate as compared to anatase TiO2 (Table 7, no. 3 and 5). The conversion rate is correlated to the surface area of the catalyst where anatase TiO2 has a larger surface area than that of Degussa P25. The results showed that the formation of surface complexes is the key point for the photocatalysis reaction to proceed under visible light irradiation. The surface complexes are formed by the interaction between the substrate and the active species (metal active sites113 or surface hydroxyl114,115). The substrate which has a higher energy level than the valance band of TiO2 acts as a donor. This is the reason that TiO2 could respond to visible light activation and promote electron transfer to the conduction band of TiO2.110 The principle of photocatalysis for TiO2 in visible light is depicted in Fig. 8.
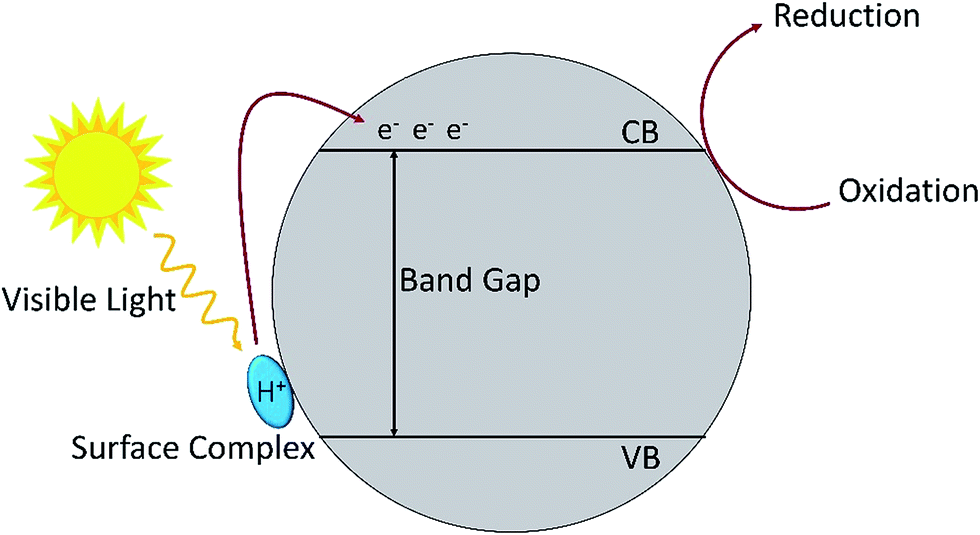 | ||
| Fig. 8 The mechanism for surface complex excitation by visible light to produce an oxidized substrate and to transfer an electron in the conduction band. | ||
5. Conclusions
TiO2 is a promising reducible oxide catalyst for green organic synthesis such as in deoxygenation, hydrogenation, esterification, transesterification, the WGS reaction, visible light-induced organic transformation etc. This catalyst can be modified by a variety of metal or non-metal dopants to increase the textural properties and thermal stability of TiO2, thereby enhancing the catalytic stability and activity. The redox and surface properties of TiO2 are strongly affected by interaction with the metal/metal oxide dopant. This metal-support interaction induced the SMSI effect during the reduction treatment by H2 and thus contributed to the high catalytic activity towards green organic synthesis.The catalytic activities of the TiO2-based catalyst are mainly influenced by the active metal dopant, where each metal has its own activity and selectivity profile for different organic synthesis reactions. Noble metals (Pt, Ru, Pd, Rh, Au) supported on TiO2 showed superior performance and high selectivity toward hydrodeoxygenation and hydrogenation. Among the noble metals, Ru/TiO2 showed a very high activity even at a very low temperature. As for the WGS reaction, the reaction activity follows the order of Pt > Rh > Ru > Pd. Ni/TiO2 is another potential non-noble catalyst extensively used in hydrogenation due to the high reactivity and cost effectiveness. The high performance of the TiO2-supported catalysts strongly relies on a higher dispersion and stabilization of small metallic particles on the TiO2 support. This is because of the synergistic effect between the bifunctional system that is generated.
The TiO2 polymorphs (anatase, rutile, and brookite) with different physicochemical properties are another key factor to influence the reactivity of organic synthesis. The existence of either one of the TiO2 polymorphs affects the performance and the TiO2 catalytic activity. Rutile has better lattice matching and interaction with Pt, Ru, and Ni if compared to the anatase phase, thereby the rutile phase TiO2 reveals a greater catalytic activity. Besides, the morphology of the TiO2 catalyst influences the conversion and product selectivity of the organic synthesis. In addition, a different type of acid site and acid density also contributed to the performance of the catalyst in the reaction. Hence, it is important to choose a synthesis method with a specific morphology to obtain a good catalytic performance.
Acknowledgements
The authors gratefully acknowledge the support of Malaysia’s Fundamental Research Grant Scheme (FP054-2013B), SATU Joint Research Scheme (RU020A-2015), University Malaya Research Grant (UMRG RP025A/RP025B/RP025C-14AET), Research University Grants (RU018D-2016), IPPP Postgraduate Research Grant (PG071-2014B) and University of Malaya Fellowship Scheme.References
- K. M. Kummer, E. Taylor and T. J. Webster, Nanosci. Nanotechnol. Lett., 2012, 4, 483–493 CrossRef CAS.
- E. A. Rozhkova, I. Ulasov, B. Lai, N. M. Dimitrijevic, M. S. Lesniak and T. Rajh, Nano Lett., 2009, 9, 3337–3342 CrossRef CAS PubMed.
- C. Lee, C. Hong, H. Kim, J. Kang and H. M. Zheng, Photochem. Photobiol., 2010, 86, 981–989 CrossRef CAS PubMed.
- C. Li, K. Sivaranjani and J. M. Kim, Catal. Today, 2016, 265, 45–51 CrossRef CAS.
- R. C. Nelson, B. Baek, P. Ruiz, B. Goundie, A. Brooks, M. C. Wheeler, B. G. Frederick, L. C. Grabow and R. N. Austin, ACS Catal., 2015, 5, 6509–6523 CrossRef CAS.
- M. Boronat and A. Corma, Langmuir, 2010, 26, 16607–16614 CrossRef CAS PubMed.
- K. Yang, Y. Zhang, Y. Li, P. Huang, X. Chen, W. Dai and X. Fu, Appl. Catal., B, 2016, 183, 206–215 CrossRef CAS.
- K. A. Davis, J. Chem. Educ., 1982, 59, 158 CrossRef CAS.
- J. Yu, L. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 CAS.
- Z. Wu, W. Zhang, F. Xiong, Q. Yuan, Y. Jin, J. Yang and W. Huang, Phys. Chem. Chem. Phys., 2014, 16, 7051–7057 RSC.
- G. I. N. Waterhouse, A. K. Wahab, M. Al-Oufi, V. Jovic, D. H. Anjum, D. Sun-Waterhouse, J. Llorca and H. Idriss, Sci. Rep., 2013, 3, 2849 CAS.
- C. G. Silva and J. L. Faria, ChemSusChem, 2010, 3, 609–618 CrossRef CAS PubMed.
- X. Shen, L. Zhu, C. Huang, H. Tang, Z. Yu and F. Deng, J. Mater. Chem., 2009, 19, 4843–4851 RSC.
- R. Giovannetti, C. A. D. Amato, M. Zannotti, E. Rommozzi, R. Gunnella, M. Minicucci and A. Di Cicco, Sci. Rep., 2015, 5, 17801 CrossRef CAS PubMed.
- H. Barndõk, D. Hermosilla, C. Han, D. D. Dionysiou, C. Negro and T. Blanco, Appl. Catal., B, 2016, 180, 44–52 CrossRef.
- S. Ma, Y. Lan, G. M. J. Perez, S. Moniri and P. J. A. Kenis, ChemSusChem, 2014, 7, 866–874 CrossRef CAS PubMed.
- H. Zhang and J. F. Banfield, J. Phys. Chem. B, 2000, 104, 3481–3487 CrossRef CAS.
- K. Bourikas, C. Kordulis and A. Lycourghiotis, Chem. Rev., 2014, 114, 9754–9823 CrossRef CAS PubMed.
- O. Carp, C. L. Huisman and A. Reller, Prog. Solid State Chem., 2004, 32, 33–177 CrossRef CAS.
- D. H. Hanaor and C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- M. Matsui and M. Akaogi, Mol. Simul., 1991, 6, 239–244 CrossRef.
- M. Koelsch, S. Cassaignon, J. F. Guillemoles and J. P. Jolivet, Thin Solid Films, 2002, 403–404, 312–319 CrossRef CAS.
- K. Okada, N. Yamamoto, Y. Kameshima, A. Yasumori and K. J. D. MacKenzie, J. Am. Ceram. Soc., 2001, 84, 1591–1596 CrossRef CAS.
- Y. G. Guo, Y. S. Hu, W. Sigle and J. Maier, Adv. Mater., 2007, 19, 2087–2091 CrossRef CAS.
- M. Hirano, C. Nakahara, K. Ota, O. Tanaike and M. Inagaki, J. Solid State Chem., 2003, 170, 39–47 CrossRef CAS.
- Y.-F. Chen, C.-Y. Lee, M.-Y. Yeng and H.-T. Chiu, J. Cryst. Growth, 2003, 247, 363–370 CrossRef CAS.
- P. S. Ha, H.-J. Youn, H. S. Jung, K. S. Hong, Y. H. Park and K. H. Ko, J. Colloid Interface Sci., 2000, 223, 16–20 CrossRef CAS PubMed.
- P. I. Gouma and M. J. Mills, J. Am. Ceram. Soc., 2001, 84, 619–622 CrossRef CAS.
- J. Porter, Y.-G. Li and C. Chan, J. Mater. Sci., 1999, 34, 1523–1531 CrossRef CAS.
- T. Mukoyama, N. Shimoda and S. Satokawa, Fuel Process. Technol., 2015, 131, 117–124 CrossRef CAS.
- K. Yamaguchi, C. Yoshida, S. Uchida and N. Mizuno, J. Am. Chem. Soc., 2005, 127, 530–531 CrossRef CAS PubMed.
- L. He, J.-Q. Wang, Y. Gong, Y.-M. Liu, Y. Cao, H.-Y. He and K.-N. Fan, Angew. Chem., 2011, 123, 10398–10402 CrossRef.
- P. D. Kent, J. E. Mondloch and R. G. Finke, J. Am. Chem. Soc., 2014, 136, 1930–1941 CrossRef CAS PubMed.
- K. G. Azzam, I. V. Babich, K. Seshan and L. Lefferts, J. Catal., 2007, 251, 163–171 CrossRef CAS.
- K. Hengst, M. Arend, R. Pfützenreuter and W. F. Hoelderich, Appl. Catal., B, 2015, 174–175, 383–394 CrossRef CAS.
- J. Zhang, Y. Li, Y. Zhang, M. Chen, L. Wang, C. Zhang and H. He, Sci. Rep., 2015, 5, 12950 CrossRef CAS PubMed.
- W. Luo, M. Sankar, A. M. Beale, Q. He, C. J. Kiely, P. C. A. Bruijnincx and B. M. Weckhuysen, Nat. Commun., 2015, 6, 6540 CrossRef PubMed.
- A. Saadi, R. Merabti, Z. Rassoul and M. M. Bettahar, J. Mol. Catal. A: Chem., 2006, 253, 79–85 CrossRef CAS.
- A. R. Ardiyanti, A. Gutierrez, M. L. Honkela, A. O. I. Krause and H. J. Heeres, Appl. Catal., A, 2011, 407, 56–66 CrossRef CAS.
- P. i. Mäki-Arvela, B. Rozmysłowicz, S. Lestari, O. Simakova, K. Eränen, T. Salmi and D. Y. Murzin, Energy Fuels, 2011, 25, 2815–2825 CrossRef.
- Y. Yan, J. Miao, Z. Yang, F.-X. Xiao, H. B. Yang, B. Liu and Y. Yang, Chem. Soc. Rev., 2015, 44, 3295–3346 RSC.
- H. G. Manyar, C. Paun, R. Pilus, D. W. Rooney, J. M. Thompson and C. Hardacre, Chem. Commun., 2010, 46, 6279–6281 RSC.
- D. Haffad, U. Kameswari, M. M. Bettahar, A. Chambellan and J. C. Lavalley, J. Catal., 1997, 172, 85–92 CrossRef CAS.
- R. Palcheva, L. Dimitrov, G. Tyuliev, A. Spojakina and K. Jiratova, Appl. Surf. Sci., 2013, 265, 309–316 CrossRef CAS.
- E. W. Zhao, H. Zheng, K. Ludden, Y. Xin, H. E. Hagelin-Weaver and C. R. Bowers, ACS Catal., 2016, 974–978, DOI:10.1021/acscatal.5b02632.
- J. C. Colmenares, P. Lisowski, D. Łomot, O. Chernyayeva and D. Lisovytskiy, ChemSusChem, 2015, 8, 1676–1685 CrossRef CAS PubMed.
- S. J. Tauster, S. C. Fung and R. L. Garten, J. Am. Chem. Soc., 1978, 100, 170–175 CrossRef CAS.
- Y.-G. Wang, Y. Yoon, V.-A. Glezakou, J. Li and R. Rousseau, J. Am. Chem. Soc., 2013, 135, 10673–10683 CrossRef CAS PubMed.
- Y. Xu, J. Ma, Y. Xu, H. Li, H. Li, P. Li and X. Zhou, Appl. Catal., A, 2012, 413–414, 350–357 CrossRef CAS.
- L. P. Matte, A. S. Kilian, L. Luza, M. C. M. Alves, J. Morais, D. L. Baptista, J. Dupont and F. Bernardi, J. Phys. Chem. C, 2015, 119, 26459–26470 CAS.
- L. Torrente-Murciano, A. A. Lapkin, D. V. Bavykin, F. C. Walsh and K. Wilson, J. Catal., 2007, 245, 272–278 CrossRef CAS.
- C. Newman, X. Zhou, B. Goundie, I. T. Ghampson, R. A. Pollock, Z. Ross, M. C. Wheeler, R. W. Meulenberg, R. N. Austin and B. G. Frederick, Appl. Catal., A, 2014, 477, 64–74 CrossRef CAS.
- M. Jakob, H. Levanon and P. V. Kamat, Nano Lett., 2003, 3, 353–358 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- C. Sun and S. C. Smith, J. Phys. Chem. C, 2012, 116, 3524–3531 CAS.
- M. J. Tillotson, P. M. Brett, R. A. Bennett and R. Grau-Crespo, Surf. Sci., 2015, 632, 142–153 CrossRef CAS.
- J. Fang, J. Li, B. Zhang, X. Yuan, H. Asakura, T. Tanaka, K. Teramura, J. Xie and N. Yan, Nanoscale, 2015, 7, 6325–6333 RSC.
- S.-C. Qi, X.-Y. Wei, Z.-M. Zong and Y.-K. Wang, RSC Adv., 2013, 3, 14219–14232 RSC.
- Y. Zhu, D. Liu and M. Meng, Chem. Commun., 2014, 50, 6049–6051 RSC.
- M. E. Manríquez, T. López, R. Gómez and J. Navarrete, J. Mol. Catal. A: Chem., 2004, 220, 229–237 CrossRef.
- R. E. Olsen, C. H. Bartholomew, B. Huang, C. Simmons and B. F. Woodfield, Microporous Mesoporous Mater., 2014, 184, 7–14 CrossRef CAS.
- P. Periyat, K. V. Baiju, P. Mukundan, P. K. Pillai and K. G. K. Warrier, Appl. Catal., A, 2008, 349, 13–19 CrossRef CAS.
- B. M. Reddy, I. Ganesh and A. Khan, J. Mol. Catal. A: Chem., 2004, 223, 295–304 CrossRef CAS.
- G. López-Granada, J. D. O. Barceinas-Sánchez, R. López and R. Gómez, J. Hazard. Mater., 2013, 263, 84–92 CrossRef PubMed.
- M. Nolan, J. Chem. Phys., 2013, 139, 184710 CrossRef PubMed.
- Q. Bu, H. Lei, A. H. Zacher, L. Wang, S. Ren, J. Liang, Y. Wei, Y. Liu, J. Tang, Q. Zhang and R. Ruan, Bioresour. Technol., 2012, 124, 470–477 CrossRef CAS PubMed.
- M. Snåre, I. Kubičková, P. Mäki-Arvela, K. Eränen and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- L. Nie and D. E. Resasco, J. Catal., 2014, 317, 22–29 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Delichère and C. Geantet, Appl. Catal., B, 2011, 101, 246–255 CrossRef CAS.
- D. Kubička, J. Horáček, M. Setnička, R. Bulánek, A. Zukal and I. Kubičková, Appl. Catal., B, 2014, 145, 101–107 CrossRef.
- J. Wildschut, F. H. Mahfud, R. H. Venderbosch and H. J. Heeres, Ind. Eng. Chem. Res., 2009, 48, 10324–10334 CrossRef CAS.
- L. He, C. Wu, H. Cheng, Y. Yu and F. Zhao, Catal. Sci. Technol., 2012, 2, 1328–1331 CAS.
- L. Cao, Z. Gao, S. L. Suib, T. N. Obee, S. O. Hay and J. D. Freihaut, J. Catal., 2000, 196, 253–261 CrossRef CAS.
- V. N. Bui, D. Laurenti, P. Afanasiev and C. Geantet, Appl. Catal., B, 2011, 101, 239–245 CrossRef CAS.
- B. S. Gevert, J. E. Otterstedt and F. E. Massoth, Appl. Catal., 1987, 31, 119–131 CrossRef CAS.
- A. F. H. Studentschnig, S. Schober and M. Mittelbach, Energy Fuels, 2013, 27, 7480–7484 CrossRef CAS.
- T. Omotoso, S. Boonyasuwat and S. P. Crossley, Green Chem., 2014, 16, 645–652 RSC.
- J. M. G. Carballo, J. Yang, A. Holmen, S. García-Rodríguez, S. Rojas, M. Ojeda and J. L. G. Fierro, J. Catal., 2011, 284, 102–108 CrossRef CAS.
- J. E. Rekoske and M. A. Barteau, J. Phys. Chem. B, 1997, 101, 1113–1124 CrossRef CAS.
- R. I. Bickley, T. Gonzalez-Carreno, J. S. Lees, L. Palmisano and R. J. D. Tilley, J. Solid State Chem., 1991, 92, 178–190 CrossRef CAS.
- B. M. Xaba and J. P. R. de Villiers, Ind. Eng. Chem. Res., 2016, 55, 9397–9407 CrossRef CAS.
- J. H. Kang, E. W. Shin, W. J. Kim, J. D. Park and S. H. Moon, J. Catal., 2002, 208, 310–320 CrossRef CAS.
- Y. Li, Y. Fan, H. Yang, B. Xu, L. Feng, M. Yang and Y. Chen, Chem. Phys. Lett., 2003, 372, 160–165 CrossRef CAS.
- J. A. Toledo-Antonio, C. Ángeles-Chávez, M. A. Cortés-Jácome, I. Cuauhtémoc-López, E. López-Salinas, M. Pérez-Luna and G. Ferrat-Torres, Appl. Catal., A, 2012, 437–438, 155–165 CrossRef CAS.
- K. J. A. Raj, M. G. Prakash, R. Mahalakshmy, T. Elangovan and B. Viswanathan, Catal. Sci. Technol., 2012, 2, 1429–1436 CAS.
- V. Nichele, M. Signoretto, F. Menegazzo, I. Rossetti and G. Cruciani, Int. J. Hydrogen Energy, 2014, 39, 4252–4258 CrossRef CAS.
- R. K. Dhokale, H. M. Yadav, S. N. Achary and S. D. Delekar, Appl. Surf. Sci., 2014, 303, 168–174 CrossRef CAS.
- N. Yao, J. Chen, J. Zhang and J. Zhang, Catal. Commun., 2008, 9, 1510–1516 CrossRef CAS.
- J. Chen, N. Yao, R. Wang and J. Zhang, Chem. Eng. J., 2009, 148, 164–172 CrossRef CAS.
- C.-H. Han, D.-W. Hong, I.-J. Kim, J. Gwak, S.-D. Han and K. C. Singh, Sens. Actuators, B, 2007, 128, 320–325 CrossRef CAS.
- K. Fan, J. Chen, F. Yang and T. Peng, J. Mater. Chem., 2012, 22, 4681–4686 RSC.
- J. Fu, X. Lu and P. E. Savage, Energy Environ. Sci., 2010, 3, 311–317 CAS.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- C.-H. Lin, S.-H. Chien, J.-H. Chao, C.-Y. Sheu, Y.-C. Cheng, Y.-J. Huang and C.-H. Tsai, Catal. Lett., 2002, 80, 153–159 CrossRef CAS.
- Z. Li, R. Wnetrzak, W. Kwapinski and J. J. Leahy, ACS Appl. Mater. Interfaces, 2012, 4, 4499–4505 CAS.
- R. M. de Almeida, L. K. Noda, N. S. Gonçalves, S. M. P. Meneghetti and M. R. Meneghetti, Appl. Catal., A, 2008, 347, 100–105 CrossRef CAS.
- E. Taarning, I. S. Nielsen, K. Egeblad, R. Madsen and C. H. Christensen, ChemSusChem, 2008, 1, 75–78 CrossRef CAS PubMed.
- A. Corma, Chem. Rev., 1995, 95, 559–614 CrossRef CAS.
- K. G. Azzam, I. V. Babich, K. Seshan and L. Lefferts, Appl. Catal., A, 2008, 338, 66–71 CrossRef CAS.
- G. Jacobs, L. Williams, U. Graham, D. Sparks and B. H. Davis, J. Phys. Chem. B, 2003, 107, 10398–10404 CrossRef CAS.
- A. G. Chakinala, D. W. F. Brilman, W. P. M. van Swaaij and S. R. A. Kersten, Ind. Eng. Chem. Res., 2010, 49, 1113–1122 CrossRef CAS.
- P. Panagiotopoulou and D. I. Kondarides, J. Catal., 2004, 225, 327–336 CrossRef CAS.
- H. Iida and A. Igarashi, Appl. Catal., A, 2006, 298, 152–160 CrossRef CAS.
- I. D. González, R. M. Navarro, M. C. Álvarez-Galván, F. Rosa and J. L. G. Fierro, Catal. Commun., 2008, 9, 1759–1765 CrossRef.
- M. Calatayud, A. Markovits, M. Menetrey, B. Mguig and C. Minot, Catal. Today, 2003, 85, 125–143 CrossRef CAS.
- K. G. Azzam, I. V. Babich, K. Seshan and L. Lefferts, J. Catal., 2007, 251, 153–162 CrossRef CAS.
- C. Hernandez-Mejia, E. S. Gnanakumar, A. Olivos-Suarez, J. Gascon, H. F. Greer, W. Zhou, G. Rothenberg and N. Raveendran Shiju, Catal. Sci. Technol., 2016, 6, 577–582 CAS.
- J. Chen, J. Cen, X. Xu and X. Li, Catal. Sci. Technol., 2016, 6, 349–362 CAS.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC.
- S. Yurdakal, G. Palmisano, V. Loddo, V. Augugliaro and L. Palmisano, J. Am. Chem. Soc., 2008, 130, 1568–1569 CrossRef CAS PubMed.
- N. Li, X. Lang, W. Ma, H. Ji, C. Chen and J. Zhao, Chem. Commun., 2013, 49, 5034–5036 RSC.
- X. Lang, W. Ma, Y. Zhao, C. Chen, H. Ji and J. Zhao, Chem.–Eur. J., 2012, 18, 2624–2631 CrossRef CAS PubMed.
- S. Higashimoto, N. Suetsugu, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2010, 274, 76–83 CrossRef CAS.
- S. Higashimoto, N. Kitao, N. Yoshida, T. Sakura, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2009, 266, 279–285 CrossRef CAS.
- S. Kitano, A. Tanaka, K. Hashimoto and H. Kominami, Phys. Chem. Chem. Phys., 2014, 16, 12554–12559 RSC.
| This journal is © The Royal Society of Chemistry 2016 |