
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bis(β-diketonato)- and allyl-(β-diketonato)-palladium(II) complexes: synthesis, characterization and MOCVD application†
K.
Assim
a,
M.
Melzer
b,
M.
Korb
a,
T.
Rüffer
a,
A.
Jakob
a,
J.
Noll
a,
C.
Georgi
bc,
S. E.
Schulz
bc and
H.
Lang
*a
aTechnische Universität Chemnitz, Faculty of Natural Sciences, Institute of Chemistry, Inorganic Chemistry, 09107 Chemnitz, Germany. E-mail: heinrich.lang@chemie.tu-chemnitz.de
bTechnische Universität Chemnitz, Center for Microtechnologies, 09107 Chemnitz, Germany
cFraunhofer Institute for Electronic Nano Systems (ENAS), Technologie-Campus 3, 09126 Chemnitz, Germany
First published on 27th October 2016
Abstract
The syntheses and characterization of the palladium complexes [Pd(accp)2] (7), [Pd(acch)2] (8), [Pd(η3-CH2CMeCH2)(accp)] (11), [Pd(η3-CH2CMeCH2)(acch)] (12), [Pd(η3-CH2CtBuCH2)(accp)] (13) and [Pd(η3-CH2CtBuCH2)(acch)] (14) (accp = 2-acetylcyclopentanoate; acch = 2-acetylcyclohexanoate) are reported. These complexes are available by the reaction of Haccp (2-acetylcyclopentanone) and Hacch (2-acetylcyclohexanone) with Na2[Pd2Cl6] forming 7 and 8 or with [(Pd(η3-CH2CRCH2)(μ-Cl))2] (9, R = Me; 10, R = tBu) forming 11–14. The molecular structures of 7, 8 and 14 are discussed. Complexes 7 and 8 consist of a square-planar coordinated Pd atom with two trans-positioned bidentate β-diketonate ligands. The asymmetric unit of 14 exhibits one molecule of the palladium complex and a half molecule of water. The thermal behavior of 7, 8 and 11–14 and their vapor pressure data were investigated to show, if the appropriate complexes are suited as CVD precursors for palladium layer formation. Thermogravimetric studies showed the evaporation of the complexes at atmospheric pressure upon heating. The vapor pressure of 7, 8 and 11–14 was measured by using thermogravimetric analysis, giving vapor pressure values ranging from 0.62 to 2.22 mbar at 80 °C. Chemical vapor deposition studies were carried out applying a vertical cold wall CVD reactor. Either oxygen or forming gas (N2/H2, ratio 90/10, v/v) was used as reactive gas. Substrate temperatures of 350 and 380 °C were utilized. With 11–14 dense and conformal as well as particulate palladium films were obtained, as directed by SEM studies, whereas 7 and 8 failed to give thin films, which is probably attributed to their high thermal stability in the gas phase. For all deposited layers, XPS measurements confirmed the partial oxidation of palladium to palladium(II) oxide at 380 °C, when oxygen was used as reactive gas. In contrast, thin layers of solely metallic palladium were obtained utilizing forming gas during the deposition experiments.
Introduction
Palladium-based materials are of importance in microtechnology, including their use in gas sensors, as electrode coatings and as hydrogen permeable selective membranes.1–4 Furthermore, palladium is used in homogeneous and heterogeneous catalysis for reactions including C,C cross-coupling and C–H activation, whereas various palladium species such as palladium(II) complexes, palladium films and palladium composite materials are used in this field.5–8 In terms of thin films, different methodologies have been established to produce them, for example, sputtering,9 electro-deposition,10 immersion deposition,11 Chemical Vapor Deposition (=CVD)12 and Atomic Layer Deposition (=ALD).13 In this respect, the CVD process possesses some advantages in terms of isotropic deposition, appropriate film thicknesses and conformal coverage. It further enables the coating of trenches and inner surfaces such as those found for example in zeolites.14 Hence, the deposition of palladium-based materials by CVD has attracted considerable attention, recently.4,15,16Great efforts have been made to provide suitable metal–organic and organometallic palladium compounds in order to full-fill the CVD requirements.12,17 A suitable CVD precursor should show good volatility, possess a high vapor pressure, and should undergo a straightforward and clean surface decomposition, avoiding the incorporation of impurities in the respective films.18 Moreover, CVD precursor molecules should be non-hazardous, air and moisture stable, and thermally inert during vaporization.18 The most frequently used family of palladium CVD precursors comprise η3-allyl- and β-diketonate units. In this respect, metal–organic complexes such as [Pd(η3-allyl)2] (1),19 [Pd(η3-2-Me-allyl)2] (2),19 and [Pd(acac)2] (3)20 (acac = acetylacetonate) were applied in gas phase deposition techniques, of which 1 and 2 produced high quality Pd layers containing only 1.0 wt% carbon impurity, without addition of any reactive gas.21 However, these precursors suffer from their high sensitivity against air and moisture as well as from their thermal instability during storage and evaporation.17 In contrast, bis(β-diketonato) palladium complexes as 3 are thermally stable but show low volatility and decompose only at high temperatures.17 In order to improve the CVD performance of these complexes, it has been shown that the combination of the two ligands allyl and β-diketonate is a promising approach to synthesize tailor-made precursors for Pd deposition.22–24 In the early nineties, Yuan and Puddephatt, for example, reported on the synthesis of [Pd(η3-allyl)(acac)] (4), [Pd(η3-2-Me-allyl)(acac)] (5), and [Pd(η3-2-Me-allyl)(hfac)] (6) (hfac = hexafluoroacetylacetonate) and their application in the CVD process.24 While, 4 still suffered from its poor stability, the substitution of the η3-allyl ligand in the 2-position by a methyl group, as characteristic for 5 and 6, resulted in more thermal stability. Based on these results, similar types of CVD palladium compounds were developed, including a series of Pd-β-diketoiminate- and -imino-alcoholate species.23,25 It was found that the usage of asymmetrical β-diketonate ligands provides the possibility to increase the volatility and decrease the melting point.26
Despite the so far investigated precursors, there is still need for palladium compounds containing an asymmetrical β-diketonate ligand without having halogenated groups such as CF3, which is, however, challenging since the respective metal–organic complexes only possess low vapor pressures, i.e.5 is less volatile than 6 and thus the deposition rate is five times slower under identical experimental deposition conditions.17
To counter-act this lack of halogen-free precursors, we herein present the synthesis, characterization, and CVD application of six Pd(II) species including [Pd(accp)2] (7), [Pd(acch)2] (8), [Pd(η3-CH2CMeCH2)(accp)] (11), [Pd(η3-CH2CMeCH2)(acch)] (12), [Pd(η3-CH2CtBuCH2)(accp)] (13), and [Pd(η3-CH2CtBuCH2)(acch)] (14) (accp = 2-acetylcyclopentanoate, CH3COCCO(CH2)3; acch = 2-acetylcyclohexanoate, CH3COCCO(CH2)4). Due to the asymmetrical nature of 2-acetylcyclopentanoate and 2-acetylcyclohexanoate, they are promising candidates to be used as ligands in Pd CVD application. Moreover, the applied palladium η3-bonded allyl ligands with a Me and tBu group in position 2, contribute to more thermal stability during storage and vaporization. In terms of the CVD experiments in this work, the main subjective was to investigate the deposition performance of these compounds depending on the nature of the reactive gases, oxygen and forming gas (N2/H2, 90/10, v/v).
Results and discussion
Synthesis and characterization of the palladium(II) complexes
The bis(β-diketonato)palladium(II) complexes [Pd(accp)2] (7) and [Pd(acch)2] (8) (accp = 2-acetylcyclopentanoate; acch = 2-acetylcyclohexanoate) were accessible by the reaction of Na2[Pd2Cl6]27,28 with Haccp (synthesis of 7) or Hacch (synthesis of 8) in the molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 in methanol at ambient temperature in presence of sodium carbonate (Scheme 1, Experimental section). After appropriate workup, 7 and 8 could be isolated as yellow solids in a yield of 80% (7) or virtually quantitative yield (8) (Experimental section).
:4 in methanol at ambient temperature in presence of sodium carbonate (Scheme 1, Experimental section). After appropriate workup, 7 and 8 could be isolated as yellow solids in a yield of 80% (7) or virtually quantitative yield (8) (Experimental section).
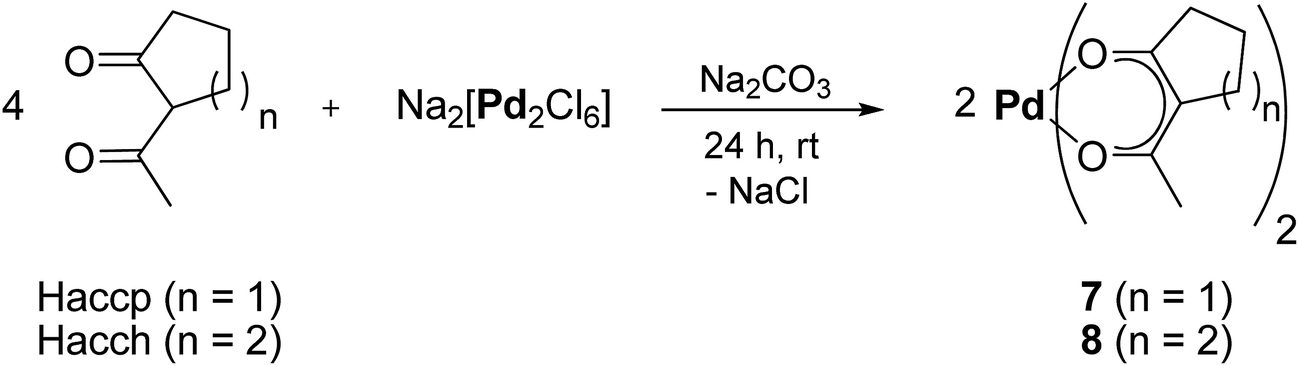 | ||
| Scheme 1 Synthesis protocol for the preparation of bis(β-diketonate)palladium(II) complexes 7 and 8 (i) methanol, ambient temperature. | ||
The synthetic methodology for the preparation of the mixed allyl-β-diketonate organometallic palladium compounds [Pd(η3-CH2CMeCH2)(accp)] (11), [Pd(η3-CH2CMeCH2)(acch)] (12), [Pd(η3-CH2CtBuCH2)(accp)] (13), and [Pd(η3-CH2CtBuCH2)(acch)] (14) is outlined in Scheme 2. According to the consecutive reaction procedure of Zhang et al.,29 at first the respective η3-allyl palladium(II) chloride dimers [(Pd(η3-CH2CRCH2)(μ-Cl))2] (9, R = Me; 10, R = tBu) were prepared. The highest yield for compound 9 was obtained, when the reaction was run in an aqueous solution.30,31 In this context, treatment of an aqueous solution containing [PdCl4]2− with 3-chloro-2-methyl-1-propene at 70 °C produced 9.29 In contrast, the synthesis of 10 included the reaction of [PdCl4]2− with 2,3,3-trimethyl-1-butene and NaOAc in glacial acetic acid at 85 °C (Experimental section).32 The thus obtained compounds 9 and 10 were than further reacted under basic conditions with Haccp or Hacch in aqueous diethyl ether solutions to give the respective title compounds 11–14. After sublimation at 40 °C at a pressure of 10−3 mbar the compounds were obtained as yellow solids in an over-all yield of 65–78% (Experimental section).
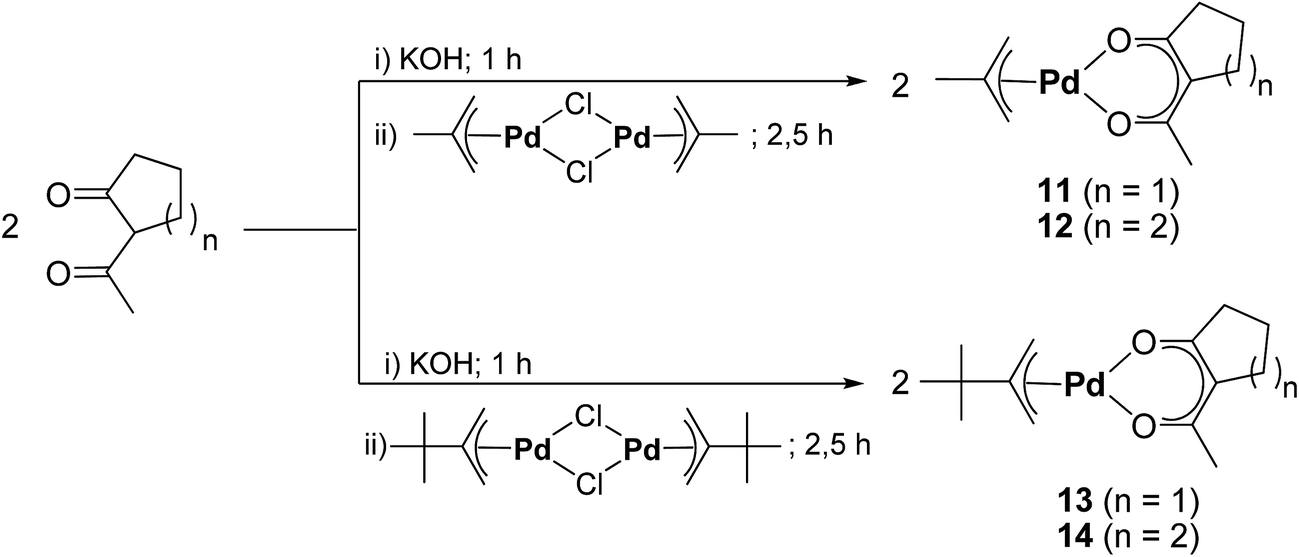 | ||
| Scheme 2 Synthesis protocol for the preparation of allyl-(β-diketonate) palladium(II) compounds 11–14 (i) distilled water, ambient temperature, 1 h stirring; (ii) aqueous diethyl ether, ambient temperature, 2.5 h stirring. | ||
Metal–organic 7 and 8 and organometallic 13 and 14 can be stored at ambient temperature in air for several days without decomposition. Nevertheless, the 2-Me-allyl derivatives 11 and 12 are somewhat more reactive and tend to decompose under similar conditions within days to give dark precipitates of unknown composition. However, when 7, 8 and 11–14 are stored at 5 °C in air they are stable for several months. These observations evince that the organic groups Me and tBu in 2 position of the allyl ligand are crucial and directly affect the thermal stability of these species, which is in accordance with the literature, where the thermal stability of similar η3-allyl bonded palladium compounds was modified by changing the substituents.17,24
All compounds are soluble in common organic solvents, including n-pentane, diethyl ether, and dichloromethane.
The identity of 7, 8 and 11–14 have been confirmed by elemental analysis, NMR (1H, 13C{1H}) and IR spectroscopy, and high resolution ESI-mass spectrometry (Experimental section). Assignments were made by 2D NMR spectroscopy. The molecular structures of 7, 8, and 14 were determined by single crystal X-ray diffraction analysis. Additionally, the thermal behavior of all compounds was investigated by thermogravimetric and vapor pressure measurements.
The 1H NMR spectra of 7 and 8 show the characteristic singlet for the Me groups at 2.09 (7) and 2.11 ppm (8), respectively. These resonances are downfield shifted when compared with the respective β-diketones (Haccp, 1.64; Hacch, 1.80 ppm), as expected. Due to the complexation of the β-diketonate to a Pd(II) ion, the signals for the hydrogen atom of Haccp (14.29 ppm) and Hacch (15.95 ppm) disappeared and thus allowed the monitoring of the reaction progress. Typical resonances of the CH2 ring protons are observed between 1.61 and 2.77 ppm (Experimental section). The 1H NMR spectrum of 8 is depicted in the ESI (Fig. SI1†). IR spectroscopy shows that the β-diketonates in 7 and 8 are chelate-bonded to Pd(II) as evidenced by the appearance of absorptions at 1571 cm−1 and 1553 cm−1, respectively.33 The coupling of ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) with ν(CC) indicates the delocalization of the negative charge within the β-diketonato unit, which is typical for this family of coordination complexes.34 Additionally, high resolution mass spectrometry showed the molecular ion peak for [M + H]+ at m/z = 357.0221 (7) and 385.0643, (8), which match the theoretical values for both 7 (m/z = 357.02) and 8 (m/z = 385.06), respectively. Furthermore, peaks associated to the ion [M + Na]+ were detected, whereas the related isotope pattern for compound 8 is shown in the ESI (Fig. SI2†).
O) with ν(CC) indicates the delocalization of the negative charge within the β-diketonato unit, which is typical for this family of coordination complexes.34 Additionally, high resolution mass spectrometry showed the molecular ion peak for [M + H]+ at m/z = 357.0221 (7) and 385.0643, (8), which match the theoretical values for both 7 (m/z = 357.02) and 8 (m/z = 385.06), respectively. Furthermore, peaks associated to the ion [M + Na]+ were detected, whereas the related isotope pattern for compound 8 is shown in the ESI (Fig. SI2†).
The syn- and anti-hydrogen CH2 allyl atoms exhibit a unique pattern in the 1H NMR spectra of 11–14 (Experimental section). While a doublet is detected for the anti protons, a further downfield shifted multiplet characterizes the protons in the syn position (Experimental section). However, under the applied conditions the fine structure of these signals could not be determined as the coupling constants between the allyl protons and their chemical shift are in the same order of magnitude. These results are quite comparable with the literature as, for example, only singlets were reported for the allyl protons in similar palladium complexes.29 In the 13C{1H} NMR spectra of 11–14 for the CH2 groups two resonance signals in a narrow range of 50.02–54.96 ppm are found, which is characteristic for η3-allyl-(β-diketonate) palladium(II) complexes.12,17,29
The IR spectra of 11–14 are characterized by frequencies associated with the ν(CO) coupled with ν(CC) vibrations, which indicates the delocalization of the negative charge, similar to 7 and 8 (vide supra).33,34 Strong bands are observed at 1596, 1572, 1627 and 1561 cm−1 for 11–14. High resolution mass spectrometry only allowed to determine [M + H]+ for 14 at m/z = 343.7612, which is in good agreement with the calculated ion peak for this complex (m/z = 343.09) (Experimental section). For 11–13 only peaks were found, which are not assignable to possible molecular fragments arising from the appropriate complexes. Furthermore, it is worth mentioning that all species 7, 8 and 11–14 are not melting but rather decompose between 100 and 130 °C, giving a black solid.
The structures of 7, 8, and 14 were determined by crystal X-ray diffraction analysis. Suitable crystals were obtained from n-pentane solutions containing 7, 8 or 14 at ambient temperature. The ORTEPs of the complexes are depicted in Fig. 1 (7, 8) and 2 (14). Relevant crystal and structural refinement data are presented in the Experimental section. Selected bond lengths and angles are given in Table 1.
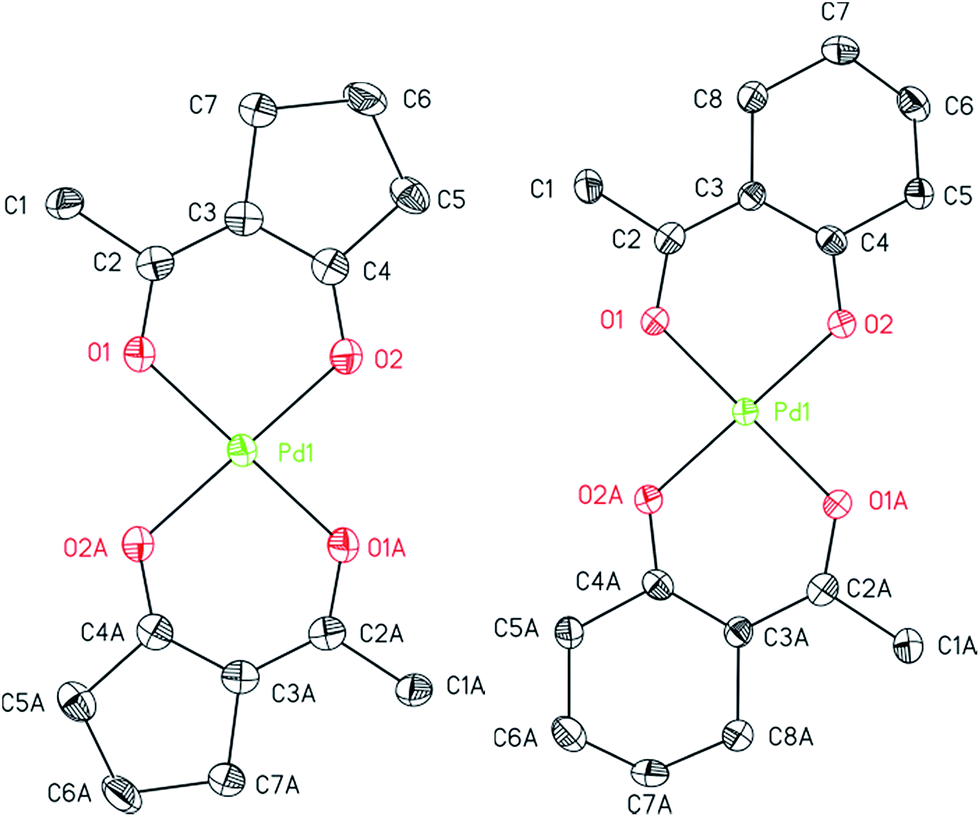 | ||
| Fig. 1 ORTEP (50% probability level) of the molecular structure of 7 (left) and 8 (right). All hydrogen atoms are omitted for clarity (symmetry transformation code for 7: A = 1 − x + 2, −y, −z + 2; for 8: A = 1 − x + 1, −y, −z + 1). | ||
| 7 | 8 | 14 | |
|---|---|---|---|
| Pd–O1 | 1.9848(19) | 1.9695(17) | 2.062(6) |
| Pd–O2 | 1.9831(18) | 1.9644(19) | 2.083(6) |
| O1–C2 | 1.284(4) | 1.282(3) | 1.256(11) |
| O2–C4 | 1.280(4) | 1.285(3) | 1.288(12) |
| C1–C2 | 1.509(7) | 1.507(4) | 1.518(13) |
| C2–C3 | 1.397(4) | 1.407(4) | 1.417(12) |
| C3–C4 | 1.397(4) | 1.397(4) | 1.383(14) |
| C4–C5 | 1.500(8) | 1.508(4) | 1.502(12) |
| Pd–C9 | — | — | 2.106(10) |
| Pd–C10 | — | — | 2.104(10) |
| Pd–C11 | — | — | 2.107(8) |
| O1–Pd–O2 | 95.52(8) | 92.58(7) | 86.7(2) |
| Pd–O1–C2 | — | — | 127.6(6) |
| Pd–O2–C4 | — | — | 128.3(6) |
| O2–Pd–C9 | — | — | 100.1(3) |
| O1–Pd–C11 | — | — | 105.2(3) |
Compound 7 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , 8 in the orthorhombic space group Pbca and 14 in the monoclinic space group C2/c. For 7 and 8, a half molecule was present in the asymmetric unit. The whole compound could be generated by C2-symmetry axis through the Pd atom and is perpendicular to the coordination plane defined by the four oxygen atoms O1, O2, O1A and O2A. The cyclopentane unit and the methyl group in 7 have been refined disordered with two sets of sights (0.75:0.25), whereby the position of the ring and the methyl group occur reversed. This also results in a trans positioning of the ligand by applying the C2 axis.
, 8 in the orthorhombic space group Pbca and 14 in the monoclinic space group C2/c. For 7 and 8, a half molecule was present in the asymmetric unit. The whole compound could be generated by C2-symmetry axis through the Pd atom and is perpendicular to the coordination plane defined by the four oxygen atoms O1, O2, O1A and O2A. The cyclopentane unit and the methyl group in 7 have been refined disordered with two sets of sights (0.75:0.25), whereby the position of the ring and the methyl group occur reversed. This also results in a trans positioning of the ligand by applying the C2 axis.
Compounds 7 and 8 consist of an ideally square-planar coordinated Pd atom with two trans-positioned bidentate β-diketonate ligands (Fig. 1). For these two species the palladium–oxygen and carbon–oxygen bond distances with 1.9644(19)–1.9848(19) and 1.280(4)–1.285(3) Å, are similar revealing delocalization of the negative charge within the β-diketonate unit (Table 1). However, the π-bonded allyl ligand in 14 results in an increase of the Pd–O bond lengths to 2.062(6)/2.083(6) Å. This affects the respective O1–Pd–O2 chelate angles that are significantly smaller in 14 (86.7(2)°) than in 7 (95.52(8)°) and 8 (92.58(7)°). The difference between the O1–Pd–O2 angles in 7 and 8 can be explained by the general geometric difference of 5-membered and 6-membered cycles present in these molecules. The structure of complexes bearing similar β-diketonates, i.e. as in [Pd(hfac)2] and trans-[Pd(btfa)2] (btfa = benzoyltrifluoro acetonate), is in good agreement with the coordination behavior reported herein.33,35
The η3-bonded tBu-allyl ligand in 14 exhibits Pd–C bond lengths of 2.104(10)–2.107(7) Å. The C9–C10–C11 angle within the allyl unit of 112.34(9)° is comparable with similar compounds, i.e. [Pd(η3-Cl(CH2)2CHCMeCH2)(acac)].36 The PdO2 plane intersects the C3 allyl plane with 71.0(6)° with the C9 and C11 atoms positioned below (C9, −0.148(13) Å; C11, −0.099(13) Å) and the C10 (0.617(12) Å) above the PdO2 plane. Thus, the tBu group occurs almost orthogonal towards the chelate metallo-cycle plane by 100.7(3)°.37
The asymmetric unit of 14 consists of one molecule of the palladium complex and a half molecule of water. The water molecule is most likely originated from the n-pentane solvent used for the crystallization process. By applying a C2 axis through O3, the whole formula unit could be calculated. Thus, the water molecule hydrogen bridges two molecules of 14 as a bond donor to the cyclohexyl bonded oxygen atoms (Fig. 2).
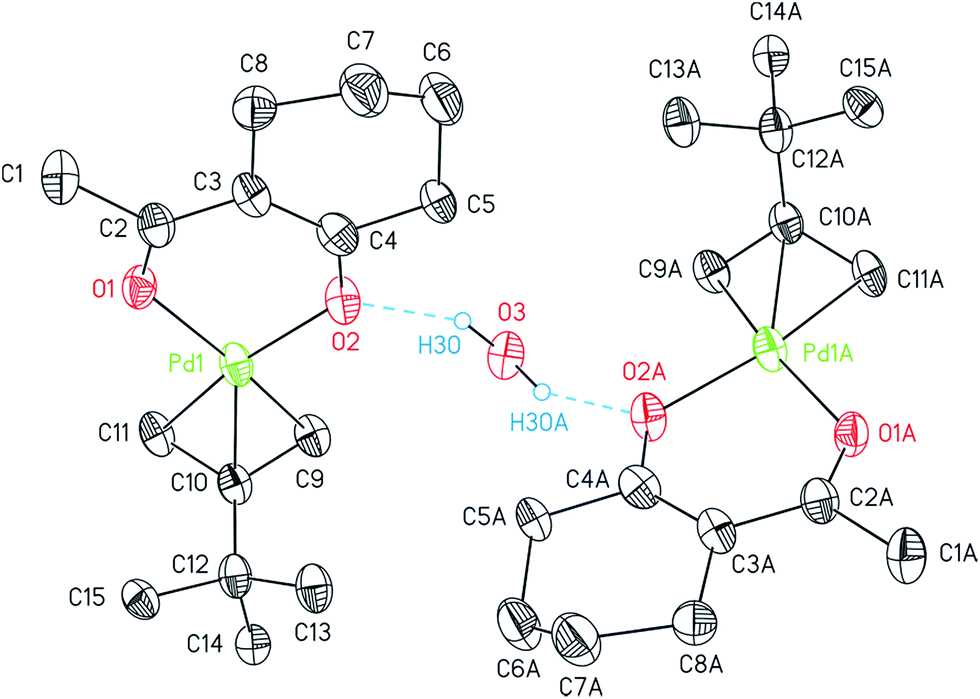 | ||
| Fig. 2 ORTEP (30% probability level) of two complexes of 14 held together by means of intermolecular hydrogen bonds with a further H2O molecule. Extension “A” refers to atoms of symmetry-generated 14 with the code 1 − x, y, 1/2 − z. All carbon-bonded hydrogen atoms are omitted for clarity. | ||
Thermal behavior
Prior to the CVD experiments the thermal decomposition behavior of 7, 8 and 11–14 was investigated by TG (=ThermoGravimetry) studies. The experiments were carried out in a nitrogen atmosphere (gas flow 20 mL min−1) as well as under oxygen (gas flow 20 mL min−1) in the temperature range of 40–800 °C with a heating rate of 10 K min−1. Additionally, a continuous nitrogen carrier gas flow of 40 mL min−1 was used for all measurements. The TG traces from the measurements in nitrogen and oxygen are depicted in Fig. 3.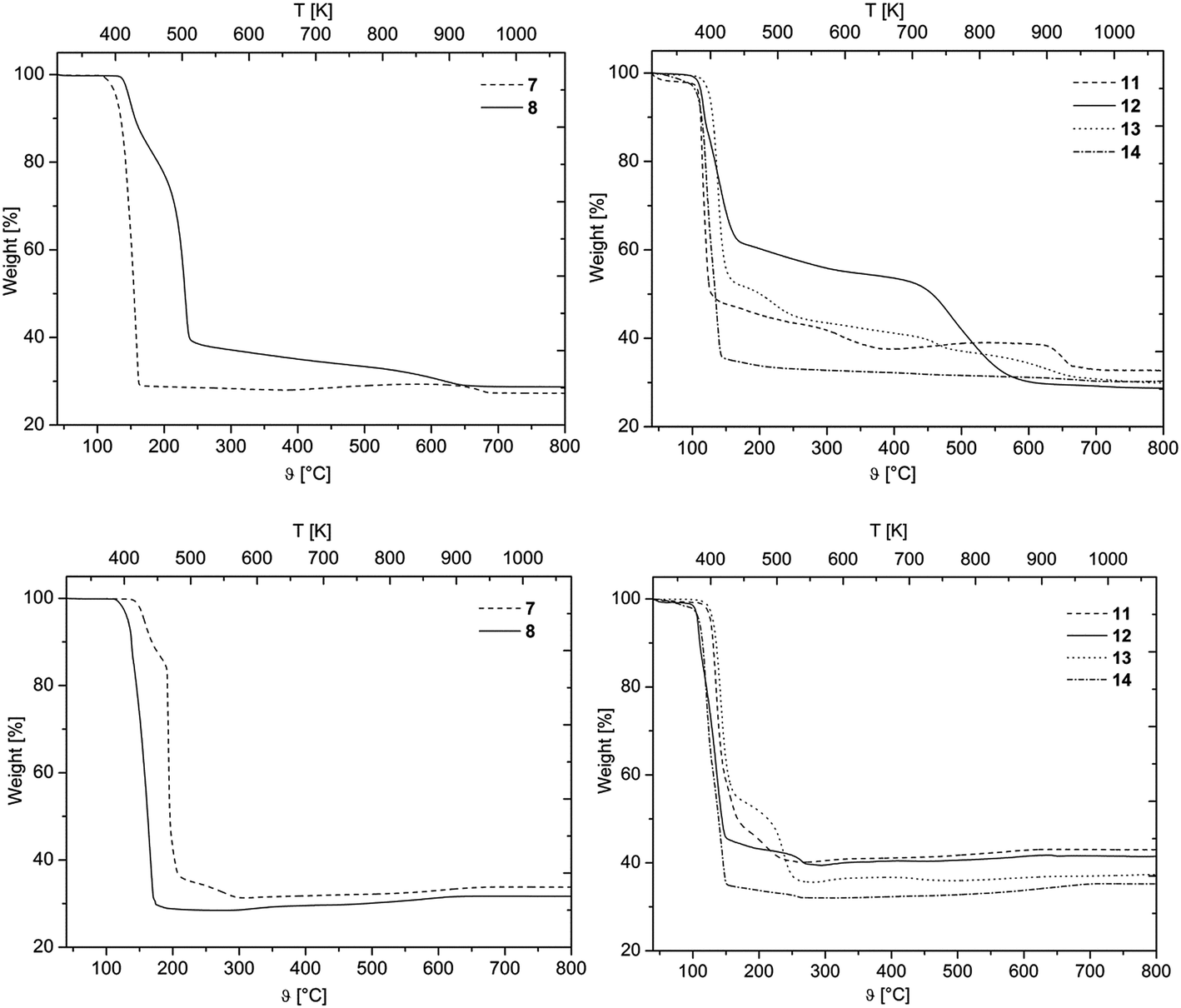 | ||
| Fig. 3 Thermogravimetric traces of 7 and 8 (left) and 11–14 (right) in nitrogen (top) and in oxygen (bottom) (gas flow, 20 mL min−1; nitrogen carrier gas flow, 40 mL min−1; heating rate 10 K min−1). | ||
As illustrated by this figure, there is no simple correlation between the thermal stability (onset temperature) and the nature of the ligands present. Under the applied conditions, all compounds undergo partial evaporation and decomposition, respectively, at atmospheric pressure as evidenced by the respective residues obtained from the experiments under nitrogen (Table 2). Since the residue value of each compound is lower in comparison to the calculated palladium content, it is presumed that 7, 8 and 11–14 are suitable for the low pressure CVD process.
| Compd | Onset temp. in N2 [°C] | Onset temp. in O2 [°C] | Calcd Pd content [%] | TG residue in N2 [%] |
|---|---|---|---|---|
| 7 | 145 | 183 | 29.3 | 28.7 |
| 8 | 113 | 145 | 27.7 | 27.2 |
| 11 | 110 | 123 | 37.3 | 32.7 |
| 12 | 102 | 101 | 35.5 | 28.8 |
| 13 | 128 | 127 | 32.4 | 29.9 |
| 14 | 110 | 112 | 31.0 | 30.1 |
The onset temperature of all compounds (under N2 and O2) is summarized in Table 2. From this table it can be seen that 7 possesses with 145 and 183 °C significantly higher onset temperatures than 8 (113 and 145 °C) under the flow of nitrogen and oxygen, respectively. Within the series of 11–14, compounds 11 and 14 behave quite similar (Table 2). Organometallic 13 exhibits the highest thermal stability of 11–14 (onset temperature 128 °C), while 12 is the compound with the lowest onset temperature of all compounds (102 °C) (Table 2). Moreover, it was found that 14 decomposes within one decomposition step, whereas 11–13 reveal several decomposition steps merging with each other (Fig. 3). For 12 and 13 the final decomposition steps occur at 450 and 650 °C, respectively. A similar thermal decomposition behavior was observed for the respective compounds if the TG studies were conducted in a stream of oxygen (Fig. 3, bottom). The onset temperatures of the compounds remain quite the same, except for 11, where the onset temperature in oxygen (123 °C) is higher than the one under nitrogen (110 °C). Additionally, it was found that the mass ratio of all samples, increases between 400 and 800 °C, which is most probably due to the conversion of palladium to palladium(II) oxide. TG-MS (=ThermoGravimetry, MS = Mass Spectrometry) experiments were carried out in order to get a deeper insight into the decomposition mechanism of 7, 8 and 11–14. However, no clear fragmentation of the complexes could be observed and the intensities of the detected masses were as low as there was no possibility of a reliable evaluation of the data.
XRPD (=X-ray powder diffraction) studies of the TG residues confirmed the formation of Pd [ICDD 03-065-2867] under a stream of nitrogen and PdO [ICDD 00-043-1024] under oxygen as carrier gas for all samples. The diffractograms belonging to the corresponding residues of 7, 8 and 11–14 are shown in the ESI (Fig. SI3–SI14†).
Vapor pressure measurements
To obtain first information about the volatility, gas phase properties and hence the suitability of 7, 8 and 11–14 as MOCVD precursors, vapor pressure measurements were carried out. All measurements were conducted at atmospheric pressure under an atmosphere of nitrogen (gas flow 40 mL min−1). According to the thermal behavior obtained from TG studies, the measuring range was adjusted for each molecule in order to avoid thermal decomposition. To minimize the measurement errors and provide reliable experimental data, each study was carried out thrice. The methodology used within this work is based on the mass-loss of the samples as a function of increasing temperature. Therefore, a TG system was applied to determine the mass-loss in an isothermal phase at different temperatures as described in ref. 38 and 39. The respective vapor pressure as a function of temperature is depicted in Fig. 4. The linear regression of the data giving the characteristic Antoine parameters for each compound according to the Antoine eqn (1) are summarized in Table 3.| logp = A − B/T | (1) |
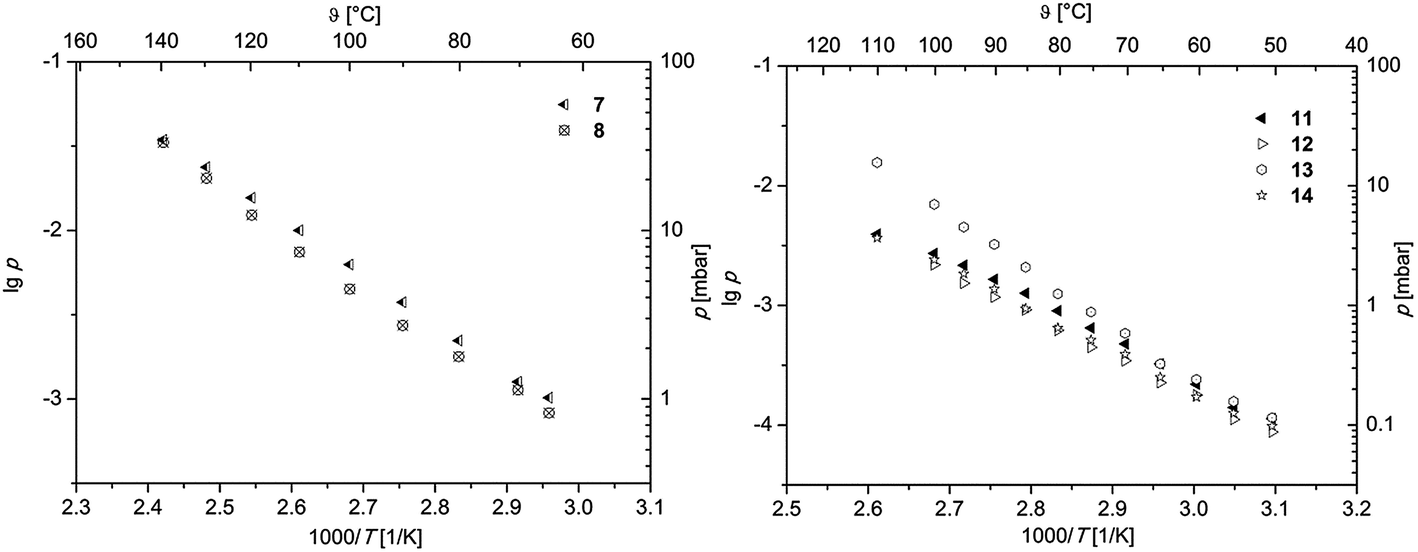 | ||
| Fig. 4 Vapor pressure of 7, 8 (left) and 11–14 (right) in an atmosphere of nitrogen (40 sccm). | ||
| logp [mbar] = (A − B/T) × 1000a |
|||||
|---|---|---|---|---|---|
| Compd | A | B | R 2 | P 80 °C [mbar] | ΔHsub (exp.) [kJ mol−1] |
| a A and B = Antoine parameters; T = temperature; R2 = coefficient of determination. | |||||
| 7 | 5.55 | 2892 | 0.999 | 2.22 | 57.46 |
| 8 | 5.59 | 2941 | 0.994 | 1.78 | 56.31 |
| 11 | 6.39 | 3338 | 0.993 | 0.90 | 64.07 |
| 12 | 6.45 | 3404 | 0.998 | 0.62 | 64.90 |
| 13 | 9.71 | 4432 | 0.994 | 1.25 | 85.57 |
| 14 | 6.45 | 3390 | 0.997 | 0.65 | 64.90 |
In order to ensure better comparability, the vapor pressures of all species at 80 °C are discussed (Table 3). As it can be seen from Table 3, the highest vapor pressure was found at 2.22 mbar for 7, while 8 possesses with 1.78 mbar a lower one. Although 8 shows a onset temperature of 113 °C (from TG, Table 2), its vapor pressure measurement revealed a linear dependence up to 140 °C. This indicates that the thermal decomposition of 8 does not occur at the onset temperature but rather above 140 °C. The determined vapor pressure values are comparable with similar complexes containing CF3 groups, i.e. [Pd(hfac)2] (hfac = hexafluoroacetylacetonate), which is reported to have a vapor pressure of 1.4 mbar at 60 °C.12
In comparison to the palladium bis-β-diketonates 7 and 8, it was found that the respective η3-allyl-β-diketonates 11–14 exhibit lower volatilities. Within the series 11–14, the 2-tBu-allyl containing complexes 13 and 14 reveal higher vapor pressures in comparison to those ones featuring a methyl group in 2 position as characteristic for 11 and 12. This notable difference may be explained in part by the larger tBu group compared to the Me unit, which shields the metal ion from intermolecular forces by surrounding it with a hydrocarbon shell making the respective compounds more volatile.40 The nature of the β-diketonates accp and acch provides a second trend in the volatility of these compounds. Comparison of 13 with 14 featuring either an accp or acch ligand indicates that 13 (1.25 mbar) reveals a significant higher vapor pressure than 14 (0.65 mbar), which is most probably based on the different geometric situations, i.e. 5-membered vs. 6-membered cycles. Similar observations have been made in the case of 11 and 12, where vapor pressures of 0.90 (11) and 0.62 mbar (12) were found. Comparing these values with those arising from the literature, [Pd(acac)2] (1.4 mbar at 60 °C) and [Pd(η3-allyl)(hfac)] (3.3 mbar at 100 °C), show that 11–14 exhibit similar vapor pressures.12,17 However, the lack of vapor pressure measurements for halogen-free systems such as [Pd(η3-2-Me-allyl)(acac)] and [Pd(η3-allyl)(acac)] is remarkable, although several reports claim that such palladium complexes are less volatile.17,24
Chemical vapor deposition experiments
Chemical vapor deposition experiments using 7, 8 and 11–14 as CVD precursors were performed in a home-built vertical cold-wall CVD reactor equipped with a continuous evaporation system.38 The depositions were conducted using nitrogen (gas flow 60 sccm) as carrier gas and oxygen (gas flow 40 sccm) or forming gas (gas flow 40 sccm, ratio N2:H2 = 90:10 (v/v)) as reactive gas. Hereinafter, the term CVD(O2) is used for the experiments with oxygen and CVD(N2/H2) for those with forming gas. As substrate silicon wafer with a dimension of 20 × 20 mm covered with a 100 nm thick SiO2 layer were used. The applied deposition conditions are summarized in Table 4.
| Compd | Reactive gas [sccm] | Heater θ [°C] | Film thicknessb [nm] | Growth rate [nm min−1] |
|---|---|---|---|---|
|
a Nitrogen gas flow rate 40 sccm, oxygen gas flow rate 40 sccm, forming gas (ratio N2:H2 = 90:10 (v/v)) flow rate 40 sccm, pressure 0.8 mbar.
b Determined by cross-sectional images.
|
||||
| 11 | O2 | 380 | 19 | 1.3 |
| 11 | N2/H2 | 350 | 30 | 2.0 |
| 12 | O2 | 380 | 28 | 1.9 |
| 12 | N2/H2 | 350 | 59 | 3.9 |
| 13 | O2 | 380 | 15 | 1.0 |
| 13 | N2/H2 | 350 | 35 | 2.3 |
| 14 | O2 | 380 | 27 | 1.8 |
| 14 | N2/H2 | 350 | 36 | 2.4 |
Under the applied deposition conditions, the two bis(β-diketonato)palladium(II) complexes 7 and 8 failed to give thin layers, which can be explained by the higher thermal stability of these molecules in the gas phase, when compared with η3-allyl-β-diketonates 11–14. This can be explained by the higher thermal stability of these compounds in the gas phase in comparison to the solid state: for example, it is reported that [Pd(hfac)2] exhibits a higher thermal stability in the gas phase than [Pd(acac)2], although [Pd(acac)2] is thermally more stabile in the solid state.41 According to this reverse order of thermal behavior, it is quite possible that the thermal stability of 7 and 8 in the gas phase is significantly higher and hence no film formation occurs.
In contrast, low pressure CVD experiments of palladium η3-allyl-β-diketonates 11–14 were carried out successfully. All precursors resulted in thin film formation in the CVD(O2) as well as in the CVD(N2/H2) experiments. To ensure a better comparability, the deposition time for each precursor was set to 15 min and the evaporation cell was heated to 60 °C to achieve a convenient precursor flow rate. The applied η3-bonded allyl ligands with Me or tBu group contribute to more thermal stability and thus no decomposition was observed during the vaporization. The substrate temperature was set to 350 °C for CVD(N2/H2) and to 380 °C for the CVD(O2) experiments. Although film depositions could be carried out at lower deposition temperatures, (starting from 250 °C), the layers obtained below 350 and 380 °C, respectively, showed dark surfaces, indicating a higher carbon impurity level. This could be proven by appropriate XPS measurements and is in well agreement with investigations from the literature.24
Film characterization
The layers deposited by CVD(N2/H2) are glossy metallic, while those obtained from CVD(O2) experiments are bright blue colored. The morphology and chemical composition of the as-deposited layers were studied using Scanning Electron Microscopy (=SEM), Energy Dispersive X-ray spectroscopy (=EDX), X-ray Photoelectron Spectroscopy (=XPS) and X-ray Powder Diffraction (=XRPD) analysis.Depending on the reactive gas and the nature of the precursor, the film morphology varies (Fig. 5). As it can be seen from Fig. 5, deposits obtained from the CVD(N2/H2) experiments are not always uniform (A**–D**). For example, 14 resulted in thin films with cracks and fissures leading to a particulate structure with separated domains with an average size of 59 (±17 nm). Similar structural patterns were found for the deposits obtained from 11 and 13 as the cross-section images confirmed the formation of separated particles on the substrate (Fig. SI16 and SI20†).
 | ||
| Fig. 5 SEM images of all deposited films (applied parameters are given in Table 4). * Films obtained by CVD experiments with oxygen (CVD(O2)). ** Films obtained by CVD experiments with forming gas (CVD(N2/H2)). A: Deposits obtained from 11, B: from 12, C: from 13, D: 14, E: cross-section image of B**. | ||
In contrast, closed and homogeneous layers with a granulated topography were formed by applying the CVD(O2) experiments. In general, there is no significant optical difference in the morphology of the films obtained from 11–14 (Fig. 5, A*–D*).
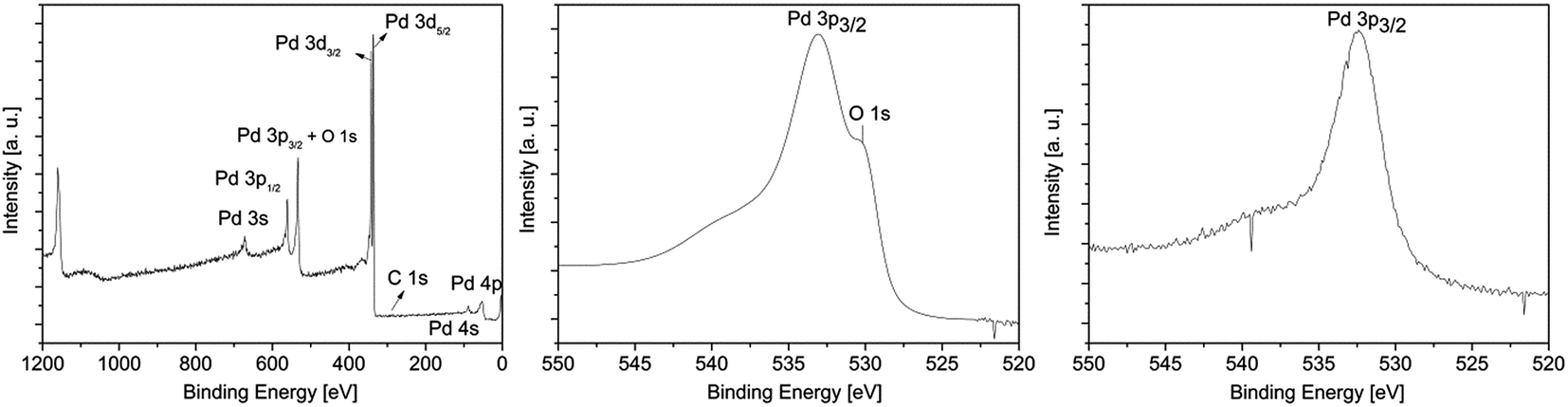 | ||
| Fig. 6 XPS survey spectrum of 13 (left); detailed XPS spectra of the Pd 3p3/2 peak obtained from the layers C* (middle) and C** (right). | ||
Cross-sectional studies were carried out for all samples in order to determine the layer thicknesses and growth rate, respectively (Table 4). Representatively, the cross-section view of the layer formed by 12 is depicted in Fig. 5, whereas all the other cross-section images are presented in the ESI (Fig. SI15–SI22†). It was found that 11–14 possess a significantly higher deposition rate in the CVD(N2/H2) experiments as in CVD(O2), which is most probably attributed to the fact that hydrogen enhances the thermal decomposition of these compounds. Gas phase analyses of similar molecules, such as [Pd(η3-allyl)(η5-C5H5)] and [Pd(acac)2] revealed that the η3-allyl as well as the β-diketonate ligands were easily hydrogenated in the gas phase enhancing the reactivity of the related molecules during the CVD process.17,41 Within the series 11–14, it is estimated that 12 has the highest reactivity, since it revealed the highest growth rate in CVD(O2) (1.9 nm min−1) as well as in CVD(N2/H2) (3.9 nm min−1) experiments. All the other compounds show similar deposition rates ranging from 1.0–1.8 nm min−1 for CVD(O2) and 2.0–2.4 nm min−1 for CVD(N2/H2).
The obtained surface morphologies of the as-deposited layers are in good agreement with those reported in the literature using palladium precursors such as [Pd(OAc)2],2 [Pd(η3-2-Me-allyl)(acac)]24 and [Pd(hfda)]23 (hfda = OC(CF3)2CH2-C(Me)NMe). Furthermore, particulate deposits such as those obtained from the CVD(N2/H2) approach are often associated with the formation of composite materials. In presence of zeolite- or metal oxide-based substrates, comparable palladium nanoparticles were deposited via the CVD process using precursors like [Pd(η3-allyl)(η5-C5H5)] and [Pd(acac)2].4,42 These composite materials are of interest in catalysis and energy storage.4,15,43
The chemical composition of the as-deposited films was investigated by EDX, showing a similar pattern of palladium signals for all samples. Comparing the films from CVD(N2/H2) and CVD(O2), a higher palladium content was found in the layers obtained by the CVD(N2/H2) experiments. In addition, elements such as silicon, oxygen and carbon were detected, whereas more oxygen was established in the layers formed by CVD(O2). The EDX spectra of all deposited layers are depicted in the ESI (Fig. SI23–SI26†). Since the EDX technique is operating with high energies (generally 3–10 keV) silicon and oxygen can be originated from the Si/SiO2 substrate.44 In order to determine the film composition without penetration of the Si/SiO2 substrate, surface sensitive ex situ XPS measurements were performed.
All XPS measurements were carried out after a sputtering process was performed to remove airborne hydrocarbon contaminations as well as carbon impurities, due to the absorbed and undecomposed precursor molecules on the topmost layers. The argon sputter gun was operated at 4 keV energy, 5 mA current and at an angle of 100° with respect to the sample plane. The sputter cleaning was applied for 2 min. For the survey spectrum, a step size of 1.0 eV was applied with a measurement time of 0.8 s per measurement point, whereas the detail spectra (Fig. 7, middle and right) were carried out with a step size of 0.1 eV and a measurement time of 1.6 s per data point. For all measurements a pass energy of 200 eV was used. The atomic composition of the samples was obtained by applying Scofield relative sensitivity factors (=RSF).45 The peaks arising from Pd 3p3/2, O 1s and Si 2s appearing at 532.4, 531.0 and 153.6 eV were used to calculate the chemical composition. In addition, the weak C 1s signal at 284.2 eV, which can be assigned to graphitic carbon, was utilized to determine the carbon contamination level.23 A typical XPS spectrum of a Pd film deposited at 350 °C using CVD precursor 13 is shown in Fig. 6. Moreover, detailed XPS spectra of the Pd 3p3/2 peak obtained from the layers C* and C** are presented in the same figure. The XPS spectra of all other samples are presented in the ESI (Fig. SI27–SI30†) and the related XPS results are summarized in Table 5.
 | ||
| Fig. 7 XRPD pattern of the layer using 13 as precursor in CVD(O2) (left) and CVD(N2/H2), (right), applied deposition parameters are given in Table 4. | ||
As it can be seen from Table 5, an oxygen content of about 25 mol% was detected for oxygen as coreactant independently from the applied precursor. In order to obtain the oxygen content of the films, the Casa XPS 2.3.16 Pre-rel 1.4 software was used for the deconvolution of the Pd 3p/O 1s area. This procedure is necessary, due to the overlapping of the Pd 3p3/2 with the O 1s peak (Fig. 6, middle). To receive the appropriate fitting parameters a metallic Pd reference sample prepared by PVD (=Physical Vapor Deposition) was analyzed by XPS, since this reference sample is oxygen free. The respective fitting parameters plus the one for the CVD samples added oxygen peak are displayed in the ESI (Fig. SI31†). It is suggested that the oxygen contamination in the layers, ranging from 25.0–26.1 mol%, are caused by PdO. This was proven by the Pd 3d5/2 peak, which is shifted between 337.0 eV for PdO and 335.0 eV for Pd.46,47 No peak is observed at 338.0 eV, which is characteristic for PdO2.48 The position of this peak correlates well with the oxygen content of the films. However, no reasonable fitting parameters were found, which would enable the deconvolution of the Pd 3d peaks. Therefore, no values are specified for the percentage of metallic palladium and palladium(II) oxide. Since the ratio between the molar concentration of oxygen and palladium is approximately 1:3 (Table 5), it is estimated that the films consist of a PdO/Pd mixture. Furthermore, by the CVD(O2) approach no carbon incorporations occurred in the respective layers, except for compound 12 (6.2 mol%). This and the fact that PdO is formed are attributed to the decomposition behavior of these precursors in an oxygen atmosphere. Similar molecules such as [Pd(η3-allyl)(fod)] and [Pd(η3-2-Me-allyl)(fod)] (fod = tBuCOCHCOC3F7) show under CVD conditions a clean decomposition giving pure metallic palladium layers and CO2 and H2O as decomposition products.29 Lowering the deposition temperature to 340 °C (from 380 °C) allowed the formation of metallic palladium films using precursors 11–14, while due to an ineffective combustion of the ligands the carbon contamination level increased. For example, the use of 14 gave thin palladium films (84.7 mol%) with a carbon content of 15.3 mol%.
Concerning the layers formed by the CVD(N2/H2) experiments, it was found that the oxygen content is significantly decreased using 10% hydrogen in the forming gas. The Pd 3p3/2 peak occurs without an overlap with the O 1s signal at 532.4 eV, confirming the formation of metallic palladium (Fig. 6, left). No further peaks at 335.0 and 338.0 eV, respectively, indicating the formation of PdO and/or PdO2, were found.
Depending on the nature of the precursors, the corresponding layers differ considerably in their carbon content. While the accp containing compounds 11 and 13 show carbon contaminations of 6.8 and 4.5 mol%, respectively, the acch featuring complexes 12 and 14 exhibit significantly higher carbon concentrations (43.3 (12), 13.4 mol% (14)). Moreover, due to the particulated morphology revealed by SEM, silicon originated from the Si/SiO2 substrate was found in the layer obtained from precursor 14.
To evaluate the crystallinity of the as-deposited films, XRPD measurements were performed. In order to obtain surface sensitive diffraction, an incident angle of 3° was applied for the X-ray beam. Due to this measurement technique, a device-specific shift was observed for all diffractograms. Similar patterns were observed for all samples obtained by using CVD(O2), whereas broad reflexes were found confirming the particulated structure of these layers. Representative, the diffractogram of the film obtained from 13 is shown in Fig. 7, whereas all the other diffractograms are presented in the ESI (Fig. SI32–SI34†). From these figures it can be seen that the crystalline part of the film mainly consists of PdO [ICDD 03-065-2867] as evidenced by the two reflexes at 33.89 and 54.76° representing the (101) and (112) orientations. Apart this, weak and broad reflexes at 40.11, 46.65, and 68.11° were found indicating the partial formation of metallic Pd [ICCD 00-043-1024]. All other reflexes could be assigned to the Si/SiO2 substrate. Concerning the layers obtained by CVD(N2/H2), only the deposit from 13 gave an appropriate diffractogram (Fig. 7, right). This is most probably due to the low carbon contamination level (4.5 mol%), which refers to a higher grade of crystallinity. The diffractogram reveals only the reflexes characteristic for Pd [ICCD 00-043-1024], whereas the one with highest intensity is observed at 40.11°. Similar diffractograms were reported as for example thin palladium films were deposited on glass using [Pd(η3-allyl)(hfac)].49 In contrast, deposits achieved from 11, 12, and 14 did not reveal any assignable reflexes, which is most likely attributed to the high carbon content preventing crystallization of Pd.
Conclusion
The synthesis of palladium complexes of type [Pd(accp)2] (7), [Pd(acch)2] (8), Pd(η3-CH2CMeCH2)(accp)] (11), [Pd(η3-CH2CMeCH2)(acch)] (12), [Pd(η3-CH2CtBuCH2)(accp)] (13), and [Pd(η3-CH2CtBuCH2)(acch)] (14) (accp = 2-acetylcyclopentanoate; acch = 2-acetylcyclohexanoate) is described and their use as MOCVD precursors for the formation of Pd and PdO layers is discussed. Complexes 7 and 8 were prepared by a “one-pot” synthetic methodology including the reaction of a Na2[Pd2Cl6] 2.85 M stock solution with sodium carbonate and the appropriate β-diketone Haccp or Hacch. The η3-allyl-β-diketonates 11–14 were accessible in a consecutive reaction procedure including the treatment of [(Pd(η3-CH2CRCH2)(μ-Cl))2] (9, R = Me; 10, R = tBu) with Haccp or Hacch.Thermogravimetric (TG) analyses showed that all compounds undergo partial decomposition upon heating but also partial evaporation is observed. Vapor pressure measurements confirmed suitable evaporation rates for all compounds, as for example 2.22 mbar at 80 °C for 7. In comparison to previously reported vapor pressures of compounds such as [Pd(η3-allyl)(hfac)] (3.3 mbar at 100 °C) and [Pd(hfac)2] (1.4 mbar at 60 °C), the reported palladium compounds exhibit comparable volatilities.17,24 CVD studies of 11–14 resulted in the formation of palladium-containing thin films by applying a vertical cold wall CVD reactor using either oxygen (=CVD(O2)) or forming gas (=CVD(N2/H2)) as reactive gas. The substrate temperatures were set to 350 (CVD(N2/H2)) and 380 °C (CVD(O2)), respectively. It was found that all precursors exhibit higher growth rates in the CVD(N2/H2) experiments, whereas 12 (3.9 nm min−1) revealed the highest one, which is in the same order of magnitude as the deposition rate reported for [Pd(η3-allyl)(hfac)] (1.14–6.6 nm min−1).12 All received films from the CVD(O2) studies were dense and coherent as proven by SEM studies. In contrast, the layers obtained from the CVD(N2/H2) procedure revealed in part a particulated morphology, which was confirmed by SEM cross-sectional experiments. The elemental composition of the as-deposited films was analyzed by EDX spectroscopy and XPS. Depositions at 380 °C in presence of oxygen led to the formation of Pd/PdO films, whereas lowering the deposition temperature to 340 °C resulted in solely metallic Pd layers with higher carbon contamination. Moreover, the use of forming gas during the CVD(N2/H2) experiments gave metallic Pd layers with minor carbon impurities except for compound 12 (carbon content: 43.3 mol%). The high carbon concentration in this case might be attributed to the acch ligand, as also 14 revealed the second highest carbon content of 13.4 mol%. The crystallinity of the as-deposited films was proven by X-ray powder diffraction measurements. The appropriate diffractograms confirmed that the crystalline part of the deposits obtained by 11–14 consists of Pd/PdO [ICDD 03-065-2867] for CVD(O2) and Pd [ICCD 00-043-1024] for CVD(N2/H2) experiments.
In conclusion, the palladium complexes 11–14 show promising properties for the generation of palladium thin layers, as they are easy to synthesize and exhibit a high thermal stability. Moreover, these molecules belong to the family of halogen-free palladium CVD precursors possessing a higher volatility in contrast to [Pd(η3-2-Me-allyl)(acac)] and [Pd(η3-allyl)(acac)].17,24 Depending on the substrate temperature and nature of the reactive gas, Pd/PdO or metallic Pd films were achieved. The morphological diversity makes the resulting layers attractive for several application fields as, for example, catalysis for C,C coupling reactions4,50 and microelectronics for carbo nanotubes field-effect transistors (CNTFETs) or graphene-based devices, where palladium can be used as contact material to carbon nanotubes or graphene layers in future micro- and nanoelectronic devices.1,51
Experimental section
Instruments and materials
The synthesis of 7, 8 and 11–14 were performed using the Schlenk flask technique under ambient conditions. Compounds 9,2910 (ref. 32) and the stock solutions of Na2[Pd2Cl6]28 were prepared by modified literature procedures. All other chemicals were used as received without further purification.The 1H NMR spectra were recorded with a Bruker Avance III 500 spectrometer operating at 500.3 MHz for 1H and at 125.8 MHz for 13C{1H} NMR in the Fourier transform mode at 298 K. The chemical shifts are reported in δ units (parts per million) downfield from tetramethylsilane with the solvent as reference signal (1H NMR, CHCl3, δ = 7.26; 13C(1H) NMR, CHCl3, δ = 77.16). The IR spectra were recorded with a FT Nicolet IR 200 instrument. High resolution mass spectra were measured with a micrO-TOF QII Bruker Daltonite workstation using Electro-Spray Ionization (ESI). TG studies were carried out with a Mettler Toledo TGA/DSC1 1100 system equipped with an UMX1 balance. Elemental analyses were conducted using a Thermo FlashAE 1112 instrument.
X-ray diffraction analysis data of 7, 8 and 14 were collected with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at 111 (7), 115 (8) and 120 (14) K. The molecular structures were solved by direct methods using SHELXS-13 (ref. 52) and refined by full-matrix least-squares procedures on F2 utilizing SHELXL-13 (ref. 53 and 54) (7 and 14) or SHELEX-97 (8).52 All non-hydrogen atoms were refined anisotropically and a riding model was employed in the treatment of the hydrogen atom positions. Graphics of the molecular structures have been created by using ORTEP.55 CCDC 1503417 (7), 1503418 (8) and 1503419 (14) contain the supplementary crystallographic data for this paper.
Chemical vapor deposition experiments were carried out in a home-built vertical cold-wall CVD reactor equipped with a continuous evaporation system (for more details see ref. 28). A heater from BACH Resistor Ceramics GmbH dimensioned with a 20 × 60 mm surface was utilized for all film depositions. Heat capacities of up to 793 K can be achieved, controlled by a Gefran 600 module, which is connected with a Pt100 thermo sensor. The gas flow of the carrier (N2) as well as that of the reactive gases (oxygen and forming gas) was controlled by MKS type 247 mass flow controllers connected to the reactor by heated copper lines. The CVD reactor was equipped with a molecular pump (EXC 120) from Edwards vacuum and the pressure within the reactor was controlled with an Edwards Active Gauge Controller (PKR 251).
The surface morphology was investigated by field-emission scanning electron microscopy (SEM) using a ZEISS Supra60 SEM microscope. Energy-dispersive X-ray (EDX) analyses were carried out utilizing a Bruker Quantax 400 system attached to the SEM. XPS measurements were examined by a PREVAC XPS system operating at ultrahigh vacuum conditions (1 × 10−9 mbar). Monochromatic Al Kα radiation (1486.6 eV) was provided using a VG Scienta MX650 X-ray source and a monochromator system. A VG Scienta EW 3000 analyzer was applied to determine the distribution of photoelectrons. The energy distribution of the photoelectrons was measured by a VG Scienta EW3000 XPS/UPS/ARPES analyzer. XRPD measurements were carried out with a STOE-STADI-P diffractometer equipped with a germanium(111) monochromator and Cu Kα radiation (λ = 0.15406 nm, 40 kV, 40 mA).
Complex 7. Haccp (0.40 mL, 3.8 mmol). Yield: 0.53 g (1.5 mmol, 80% based on Na2[Pd2Cl6]). 1H NMR (CDCl3): δ = 1.86–1.92 (m, 4H, CCH2C), 2.09 (s, 6H, CH3), 2.52–2.56 (m, 4H, CCH2), 2.60–2.63 (m, 4H, COCH2) ppm. 13C{1H} NMR (CDCl3): δ = 21.8 (CCH2C), 24.2 (CCH2), 29.7 (CH3), 37.5 (COCH2), 110.9 (C), 184.2 (CH2CO), 193.2 (CH3CO) ppm. IR (KBr): ν = 2950 (w), 2905 (w), 2854 (w), 1571 (s), 1473 (s), 1414 (w), 1375 (m), 1277 (m), 1165 (w), 1014 (w), 964 (w), 686 (w) cm−1. HRMS (ESI-TOF): calcd for C14H19O4Pd 357.03; found m/z = 356.022 [M + H]+. Anal calcd for C14H18O4Pd: C 47.14, H 5.09; found C 47.03, H 5.12.
Crystal data for 7. C14H18O4Pd, M = 356.71 g mol−1, crystal dimensions 0.40 × 0.32 × 0.22 mm, triclinic, P
, λ = 0.71073 Å, a = 7.2285(6) Å, b = 7.6427(9) Å, c = 7.6586(7) Å, α = 62.479(10)°, β = 67.121(8)°, γ = 63.114(9)°, V = 325.90(7) Å3, Z = 1, ρcalcd = 1.817 Mg m−3, μ = 1.431 mm−1, T = 110.9(2) K, θ range 3.934–24.994°, 2563 reflections collected, 1131 independent reflections (Rint = 0.0250), R1 = 0.0232, wR2 = 0.0556 (I > 2σ(I)).
Complex 8. Hacch (0.49 mL, 3.8 mmol). Yield: 0.72 g (1.9 mmol, 99% based on Na2[Pd2Cl6]). 1H NMR (CDCl3): δ = 1.58–1.67 (m, 8H, C(CH2)2C), 2.11 (s, 6H, CH3), 3.35 (t, 3JH,H = 6.4 Hz, 4H, CCH2), 2.44 (t, 3JH,H = 6.4 Hz, 4H, COCH2) ppm. 13C{1H} NMR (CDCl3): δ = 22.17 (CCH2C), 23.46 (CCH2C), 25.65 (CCH2), 27.22 (CH3), 35.23 (COCH2), 106.37 (C), 185.04 (CH2CO), 187.44 (CH3CO) ppm. IR (KBr): ν = 2942 (w), 2836 (w), 1553 (s), 1454 (s), 1364 (m), 1348 (m), 1331 (m), 1271 (m), 1170 (m), 966 (w), 825 (w), 715 (w), 693 (w) cm−1. HRMS (ESI-TOF): calcd for C16H23O4Pd 385.06; found 384.0643 [M + H]+. Anal calcd for C16H22O4Pd: C 49.95, H 5.76; found C 49.77, H 5.76.
Crystal data for 8. C16H22O4Pd, M = 384.76 g mol−1, crystal dimensions 0.22 × 0.20 × 0.18 mm, orthorhombic, Pbca, λ = 0.71073 Å, a = 14.4316(5) Å, b = 6.5451(2) Å, c = 15.6537(5) Å, V = 1478.59(8) Å3, Z = 4, ρcalcd = 1.728 Mg m−3, μ = 1.268 mm−1, T = 115 K, θ range 2.96–24.98°, 3634 reflections collected, 1296 independent reflections (Rint = 0.0215), R1 = 0.0269, wR2 = 0.0626 (I > 2σ(I)).
Compound 11. Haccp (0.16 mL, 1.3 mmol). Yield: 0.27 g (0.94 mmol, 75% based on 9). 1H NMR (CDCl3): δ = 1.77–1.89 (m, 2H, CCH2C), 2.06 (s, 3H, CH3), 2.20 (s, 3H, CH3 of 2-methylallyl), 2.37–3.48 (m, 2H, CCH2), 2.60–2.70 (m, 2H, COCH2), 2.76 (d, 2J = 3.0 Hz, 2H, anti-H, allyl-CH2), 3.66–3.67 (m, 2H, syn-H, allyl-CH2) ppm. 13C{1H} NMR (CDCl3): δ = 20.64 (CCH2C), 23.25 (CH3, allyl), 26.75 (CCH2), 30.51 (CH3), 39.30 (COCH2), 54.96 (CH2, allyl), 55.07 (CH2, allyl), 107.93 (COCCO, β-diketonate), 129.06 (CH2CCH2, allyl), 185.33 (CH2CO), 193.23 (CH3CO) ppm. IR (KBr): ν = 2951 (m), 1596 (s), 1458 (w), 1474 (m), 1396 (m), 1308 (m), 1266 (w), 1016 (w) cm−1. Anal. cald for C11H16O2Pd: C 46.09, H 5.63; found C 45.96, H 6.18.
Compound 12. Hacch (0.17 mL, 1.3 mmol). Yield: 0.30 g (0.98 mmol, 78% based on 9). 1H NMR (CDCl3): δ = 1.63–1.65 (m, 4H, C(CH2)2C), 2.08 (s, 3H, CH3), 2.19 (s, 3H, CH3 of 2-methylallyl), 2.31–2.35 (m, 2H, CCH2), 2.37–2.44 (m, 2H, COCH2), 2.70 (d, 2J = 9.5 Hz, 2H, anti-H, allyl-CH2), 3.59–3.60 (m, 2H, syn-H, allyl-CH2) ppm. 13C{1H} NMR (CDCl3): δ = 22.88 (CCH2C), 23.30 (CCH2C), 23.88 (CH3, allyl), 27.55 (CCH2), 27.89 (CH3), 37.22 (COCH2), 54.17 (CH2, allyl), 54.71 (CH2, allyl), 106.96 (COCCO, β-diketonate), 128.23 (CH2CCH2, allyl), 185.57 (CH2CO), 188.46 (CH3CO) ppm. IR (KBr): ν = 2933 (m), 2846 (w), 1572 (s), 1455 (m), 1440 (m), 1378 (s), 1336 (m), 1268 (m), 1164 (w), 1030 (w), 964 (w), 835 (w) cm−1. Anal. calcd for C12H18O2Pd: C 47.93, H 6.03; found C 47.54, H 6.03.
Compound 13. Haccp (0.16 mL, 1.3 mmol). Yield: 0.27 g (0.82 mmol, 65% based on 10). 1H NMR (CDCl3): δ = 1.25 (s, 9H, CH3 of tBu), 1.78–1.86 (m, 2H, CCH2C), 2.03 (s, 3H, CH3), 2.35–2.46 (m, 2H, CCH2), 2.57 (d, 2J = 4.1 Hz, 2H, anti-H, allyl-CH2), 2.59–2.69 (m, 2H, COCH2), 3.75–3.76 (m, 2H, syn-H, allyl-CH2) ppm. 13C{1H} NMR (CDCl3): δ = 20.57 (CCH2C), 26.81 (CH3, β-diketonate), 30.25 (CH3, tBu), 30.55 (CCH2), 34.75 ((CH3)3C, tBu), 39.32 (COCH2), 50.95 (CH2, allyl), 51.02 (CH2, allyl), 107.73 (COCCO, β-diketonate), 142.19 (CH2CCH2, allyl), 185.34 (CH2CO), 193.04 (CH3CO) ppm. IR (KBr): ν = 3027 (m), 2404 (w), 1626 (s), 1527 (s), 1417 (m), 1290 (w), 928 (w), 710 (w) cm−1. Anal. cald for C14H22O2Pd: C 51.15, H 6.75; found C 51.22, H 7.01.
Compound 14. Hacch (0.17 mL, 1.3 mmol). Yield: 0.29 g (0.84 mmol, 65% based on 10). 1H NMR (CDCl3): δ = 1.24 (s, 9H, CH3, tBu), 1.61–1.68 (m, 4H, C(CH2)2C), 2.06 (s, 3H, CH3), 2.29–2.33 (m, 2H, CCH2), 2.35–2.43 (m, 2H, COCH2), 2.51 (d, 2J = 10.6 Hz, 2H, anti-H, allyl-CH2), 3.67–3.69 (m, 2H, syn-H, allyl-CH2) ppm. 13C{1H} NMR (CDCl3): δ = 22.92 (CCH2C), 23.91 (CCH2C), 27.57 (CH3, β-diketonate), 27.96 (CCH2), 30.32 (CH3, tBu), 34.69 (C, tBu), 37.27 (COCH2), 50.02 (CH2, allyl), 50.63 (CH2, allyl), 106.75 (COCCO, β-diketonate), 141.27 (CH2CCH2, allyl), 185.49 (CH2CO), 188.65 (CH3CO) ppm. IR (KBr): ν = 2924 (m), 2855 (w), 1561 (s), 1453 (m), 1371 (s), 1349 (m), 1332 (m), 1264 (s), 1165 (m), 1029 (w), 997 (m), 865 (w), 796 (w), 699 (m), 658 (m) cm−1. HRMS (ESI-TOF): calcd for C15H25O2Pd 343.09; found m/z = 343.7612 [M + H]+. Anal. cald for C15H24O2Pd: calcd C 52.56, H 7.06; found C 52.28, H 7.22.
Crystal data for 14. C30H50O5Pd2, M = 703.50 g mol−1, crystal dimensions 0.40 × 0.10 × 0.01 mm, monoclinic, C2/c, λ = 0.71073 Å, a = 24.443(7) Å, b = 5.4443(9) Å, c = 23.838(8) Å, β = 111.49(3)°, V = 2951.7(16) Å3, Z = 4, ρcalcd = 1.255 Mg m−3, μ = 1.255 mm−1, T = 119.95(10) K, θ range 3.848–24.999°, 8771 reflections collected, 2594 independent reflections (Rint = 0.0878), R1 = 0.0786, wR2 = 0.1778 (I > 2σ(I)).
Acknowledgements
We gratefully acknowledge the Deutsche Forschungsgemeinschaft (DFG) (GRK 1215 - Materials and Concepts for Advanced Interconnects and Nanosystems) for generous financial support. M. K. thanks the Fonds der Chemischen Industrie for a PhD fellowship. This work was partly performed within the Federal Cluster of Excellence EXC 1075 MERGE Technologies for Multifunctional Lightweight Structures, supported by the German Research Foundation (DFG). Cornelia Kowol from Fraunhofer Institute for Electronic Nano Systems (ENAS) is acknowledged for measuring the SEM and EDX spectra and Dipl.-Chem. Natalia Rüffer for performing the TG analysis experiments.References
- H. Zhong, Z. Zhang, B. Chen, H. Xu, D. Yu, L. Huang and L. Peng, Nano Res., 2015, 8, 1669–1679 CrossRef CAS.
- K. Sato, M. Natsui and Y. Hasegawa, Mater. Trans., 2015, 56, 473–478 CrossRef CAS.
- E. Czerwosz, E. Kowalska, M. Kozłowski, J. Radomska, H. Wronka, M. Angiola, A. Martucci and W. Włodarski, Procedia Eng., 2014, 87, 963–966 CrossRef CAS.
- M. Zhang, Y. Gao, C. Li and C. Liang, Chin. J. Catal., 2015, 36, 588–594 CrossRef CAS.
- S. Bräse, in Organometallics in Synthesis, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013, pp. 777–1000 Search PubMed.
- P. Serp, P. Kalck and R. Feurer, Chem. Rev., 2002, 102, 3085–3128 CrossRef CAS PubMed.
- C. Gäbler, M. Korb, D. Schaarschmidt, J. Matthäus Speck, A. Hildebrandt and H. Lang, Inorg. Chem. Commun., 2015, 54, 96–99 CrossRef.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- J.-H. Yoon, B.-J. Kim and J.-S. Kim, Mater. Chem. Phys., 2012, 133, 987–991 CrossRef CAS.
- S. Shrestha and E. J. Biddinger, Electrochim. Acta, 2015, 174, 254–263 CrossRef CAS.
- Y. Wang, W. Li, W. Wang, N. Mitsuzak, W. Bao and Z. Chen, Thin Solid Films, 2015, 586, 35–40 CrossRef CAS.
- J. R. V. Garcia and T. Goto, Mater. Trans., 2003, 44, 1717–1728 CrossRef CAS.
- D. N. Goldstein and S. M. George, Appl. Phys. Lett., 2009, 95, 143106 CrossRef.
- H. O. Pierson, Fundamentals of Chemical Vapor Deposition, Elsevier, 1999 Search PubMed.
- K. Sobczak, Int. J. Thermophys., 2015, 36, 795–803 CrossRef CAS.
- S. St. John, R. W. Atkinson, K. A. Unocic, R. R. Unocic, T. A. Zawodzinski and A. B. Papandrew, ACS Catal., 2015, 5, 7015–7023 CrossRef CAS.
- J.-C. Hierso, R. Feurer and P. Kalck, Coord. Chem. Rev., 1998, 178–180, 1811–1834 CrossRef CAS.
- F. Maury, J. Phys. IV, 1995, 5, 449–463 CrossRef.
- B. Henc, P. W. Jolly, R. Salz, S. Stobbe, G. Wilke, R. Benn, R. Mynott, K. Seevogel, R. Goddard and C. Krüger, J. Organomet. Chem., 1980, 191, 449–475 CrossRef CAS.
- M. C. Gupta, Appl. Opt., 1984, 23, 3950–3953 CrossRef CAS PubMed.
- J. E. Gozum, D. M. Pollina, J. A. Jensen and G. S. Girolami, J. Am. Chem. Soc., 1988, 110, 2688–2689 CrossRef CAS.
- G. I. Zharkova, P. A. Stabnikov, I. A. Baidina, A. I. Smolentsev and S. V. Tkachev, Polyhedron, 2009, 28, 2307–2312 CrossRef CAS.
- Y.-H. Liu, Y.-C. Cheng, Y.-L. Tung, Y. Chi, Y.-L. Chen, C.-S. Liu, S.-M. Peng and G.-H. Lee, J. Mater. Chem., 2003, 13, 135–142 RSC.
- Z. Yuan and R. J. Puddephatt, Adv. Mater., 1994, 6, 51–54 CrossRef CAS.
- Y.-L. Tung, W.-C. Tseng, C.-Y. Lee, P.-F. Hsu, Y. Chi, S.-M. Peng and G.-H. Lee, Organometallics, 1999, 18, 864–869 CrossRef CAS.
- R. Yiping Zhang and R. J. Puddephatt, Chem. Vap. Deposition, 1997, 3, 81–83 CrossRef.
- V. N. Pathak, C. K. Oza, R. Gupta, R. Tiwari and S. Chaudhary, Synth. React. Inorg. Met.–Org. Chem., 2003, 33, 607–624 CrossRef CAS.
- D. A. White, J. Chem. Soc. A, 1971, 143–145 RSC.
- Y. P. Zhang, Z. Yuan and R. J. Puddephatt, Chem. Mater., 1998, 10, 2293–2300 CrossRef CAS.
- E. W. Abel, F. G. A. Stone and G. Wilkinson, in Comprehensive Organometallic Chemistry II, Elsevier, Oxford, U. K., 1995, vol. 9 Search PubMed.
- R. C. Palenik and G. J. Palenik, Synth. React. Inorg. Met.–Org. Chem., 1992, 22, 1395–1399 CrossRef CAS.
- D. P. Hruszkewycz, L. M. Guard, D. Balcells, N. Feldman, N. Hazari and M. Tilset, Organometallics, 2015, 34, 381–394 CrossRef CAS.
- S. Okeya, S. Ooi, K. Matsumoto, Y. Nakamura and S. Kawaguchi, Bull. Chem. Soc. Jpn., 1981, 54, 1085–1095 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2008 Search PubMed.
- P. A. Stabnikov, I. A. Baidina, G. I. Zharkova, I. K. Igumenov and S. V. Borisov, J. Struct. Chem., 2000, 41, 106–115 CrossRef CAS.
- M. Farra-Hake, M. F. Rettig, J. L. Williams and R. M. Wing, Organometallics, 1986, 5, 1032–1040 CrossRef CAS.
- The value is represented by the Pd1–Ct1–C12 angle. Ct1 is defined as the centroid of C9 and C11.
- A. Tuchscherer, C. Georgi, N. Roth, D. Schaarschmidt, T. Rüffer, T. Waechtler, S. E. Schulz, S. Oswald, T. Gessner and H. Lang, Eur. J. Inorg. Chem., 2012, 4867–4876 CrossRef CAS.
- C. Georgi, A. Hildebrandt, A. Tuchscherer, S. Oswald and H. Lang, Z. Anorg. Allg. Chem., 2013, 639, 2532–2535 CrossRef CAS.
- R. E. Sievers and J. E. Sadlowski, Science, 1978, 201, 217–223 CAS.
- P. P. Semyannikov, V. M. Grankin, I. K. Igumenov and A. F. Bykov, J. Phys. IV, 1995, 5, C5-205–C5-211 Search PubMed.
- F. E. Annanouch, Z. Haddi, M. Ling, F. Di Maggio, S. Vallejos, T. Vilic, Y. Zhu, T. Shujah, P. Umek, C. Bittencourt, C. Blackman and E. Llobet, ACS Appl. Mater. Interfaces, 2016, 8, 10413–10421 CAS.
- B. Zielinska, B. Michalkiewicz, E. Mijowska and R. J. Kalenczuk, Nanoscale Res. Lett., 2015, 10, 430 CrossRef PubMed.
- J. Jeschke, S. Möckel, M. Korb, T. Rüffer, K. Assim, M. Melzer, G. Herwig, C. Georgi, S. E. Schulz and H. Lang, J. Mater. Chem. C, 2016, 2, 2319–2328 RSC.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- M. C. Militello, Surf. Sci. Spectra, 1994, 3, 387 CrossRef CAS.
- M. C. Militello, Surf. Sci. Spectra, 1994, 3, 395 CrossRef CAS.
- J. F. Moulder, W. F. Stickle and P. E. Sobol, Handbook of X-ray Photoelectron Spectroscopy: A Reference Book of Standard Spectra for Identification and Interpretation of XPS Data, Physical Electronics Division, Perkin-Elmer Corporation, 1992 Search PubMed.
- J.-C. Hierso, C. Satto, R. Feurer and P. Kalck, Chem. Mater., 1996, 8, 2481–2485 CrossRef CAS.
- J. C. Hierso, R. Feurer and P. Kalck, Chem. Mater., 2000, 12, 390–399 CrossRef CAS.
- A. D. Franklin, D. B. Farmer and W. Haensch, ACS Nano, 2014, 8, 7333–7339 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, No Title SHELXL - Program for Crystal Structure Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1503417–1503419. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra22887a |
| This journal is © The Royal Society of Chemistry 2016 |