
Concentration dependent electrochemical properties and structural analysis of a simple magnesium electrolyte: magnesium bis(trifluoromethane sulfonyl)imide in diglyme†
Abstract
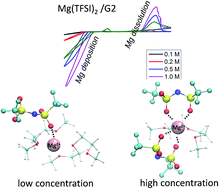
Development of Mg electrolytes that can plate/strip Mg is not trivial and remains one of the major roadblocks to advance Mg battery research. Halogen-free electrolyte has attracted great attention due to its high stability, less corrosive nature and compatibility with Mg metal anodes. However, the electrochemical properties of such electrolytes have not been analytically evaluated in the literature. Herein, we report a systematic study of the concentration-dependent electrochemical and mass transport properties of a non-aqueous, halogen-free Mg electrolyte composed of magnesium bis(trifluoromethane sulfonyl)imide in diglyme (Mg(TFSI)2/G2). Specifically, cyclic voltammograms confirm that plating and stripping of Mg in Mg(TFSI)2/G2 electrolyte occur over a wide concentration range. Results suggest a comparably difficult magnesium dissolution in Mg(TFSI)2/G2 electrolyte in contrast to in Grignard based electrolytes. Dissolution overpotential shows a non-monotonic dependence on electrolyte concentration, it requires an ∼2 V overpotential to deposit Mg. Findings also reveal concentration-dependent mass transport properties, including concentration-dependent electrolyte diffusivity and transference number. The atomic environment of the Mg(TFSI)2/G2, as being further explored by Nuclear Magnetic Resonance (NMR) measurement and Molecular Dynamics (MD) simulations, is coupled with the electrochemical measurements to explain the observed concentration-dependent mass transport properties.
 Please wait while we load your content...
Please wait while we load your content...

