
Excimer formation and dual fluorescence emission of a hydroxyquinoline based macrocyclic tripod: existence of intra and intermolecular π-stacking
Rohinia,
Minati Baralb and
B. K. Kanungo*a
aDepartment of Chemistry, Sant Longowal Institute of Engineering & Technology, Longowal, Punjab-148106, India. E-mail: b.kanungo@gmail.com
bDepartment of Chemistry, National Institute of Technology Kurukshetra, Haryana-136119, India
First published on 7th November 2016
Abstract
The excited state reaction of a conformationally flexible molecule, 1,4,7-tris-(5-methylene-8-hydroxyquinoline)-1,4,7-triazacyclononane, (9N3Me5Ox), was studied in solution. The unique geometry of this compound forces the two 8-hydroxyquinoline (8HQ) rings to undergo a face to face π–π stacking in the ground state, while the third ring forms a T-shaped orientation with face to edge interaction. The existence of intramolecular π-stacking interaction of the 8HQ units was established by 1H NMR and luminescence spectroscopy. Due to intramolecular π-stacking, chemical shifts of the 8HQ protons in neutral form were slightly shielded in the NMR spectrum. In neutral solution, excimer emission was observed with appearance of an additional strong red shifted band in the fluorescence spectra at moderate concentration due to intermolecular π-stacking interaction among the 8HQ units in the excited state. The dual emission of the neutral solution of the molecule vanished with change in pH or concentration of the solution. At very low concentration, very high concentration, as well as in low pH or high pH, only one emission band was observed. Time-resolved fluorescence data supported the nonexistence of excited state intramolecular proton transfer (ESIPT) or excited state phototautomerization. The studies of fluorescence spectra of the compound in protic, aprotic and polar protic solvent confirmed the assistance of protic solvent in formation of intermolecular π-stacking excimer. The lifetime of the tripod containing three 8HQ has been enhanced to nano-second level causing a “turn on” of fluorescence as compared to the earlier reported unsubstituted short-lived 8HQ which has life time picoseconds.
Introduction
The chemistry of 8-hydroxyquinoline (8HQ) experiences a renaissance due to 8-hydroxyquinoline (8HQ) and its derivatives have been extensively used in various fields of materials science and pharmaceutics. The wide applicability of 8HQ and its derivatives is mainly due to metal chelation properties that have been known for more than a century.1 It behaves as a bidentate ligand through N and O donor atoms, and considered as the second chelating agent in significance after EDTA.2 The fluorogenic property of 8HQ and its family, when complexed with distinct metal ions, is characterized as posing the strong potential for optoelectronic materials;3 for example, the aluminum complex of 8HQ, known as Alq3,4,5 has been applied to solar cells and optoelectronics.6 Ruthenium complexes of 8HQ are promising for catalysis in artificial photosynthetic systems for fuel production.7 8HQ and its derivatives are also famous as fluorogenic ligands for a different type of metal ions,8,9 which include rare earth metal ions,10 transition metal ions11 and lanthanides.12 They have been used analytically13 and industrially14 for the detection and separation of metal ions. Besides, they have a broad spectrum of biological activities15,16 as neuro-protection agents,17 anti-cancer agents18 and anti-HIV agents.19 Previous studies showed that, modifying an 8HQ ligand by its substituent at different positions have changed its selectivity and coordination behavior towards different metal ions.20 To improve metal binding selectivity and efficiency of 8HQ, several new polydentate chelators containing more than one 8HQ units attached to variety additional central units connecting groups at the C-2, C-3, and/or C-7 positions of 8HQ, have been synthesized.21,22 Some C3 symmetric tripodal molecules attached to the C-5 position of 8HQ have been also reported in our laboratory.23,24 In this communication, studies of a newly synthesized encapsulating polydentate macrocyclic tripod, 1,4,7-tris(5-methylene-8-hydroxyquinoline)-1,4,7-triazacyclononane, (9N3Me5Ox), (Fig. 1) are reported. | ||
| Fig. 1 Structures of 1,4,7-tris(5-methylene-8-hydroxyquinoline)-1,4,7-triazacyclononane (9N3Me5Ox). | ||
It is interesting to note that the tripod containing three units of 8HQ attached at the C-5 position to the macrocycle 9N3 displays unusual photophysical properties. In aqueous solution, and at physiological pH, the newly synthesized compound 9N3Me5Ox displays dual luminescence (Fig. 4). Interestly, with decrease or increase in pH, the dual fluorescence spectra disappear [Fig. 4(b) and (c)]. Usually, the fluorescence spectrum is characterized as a single emission band, which is independent of excitation wavelength.25 Two emission bands are rarely observed in fluorescence spectra, which may be possible due to excited state reactions, excimer or exciplex formation,26 where one ground state species creates two excited state species, thus exhibit two emission bands. Examples of such phenomena are mainly dominated by the pyrenes and their compounds containing more than one pyrene units.27–29 Very few other examples, particularly, compounds of 3-hydroxyisoquinoline (3HIQ),30 tripodal based quinoline,31 2-O-tert-butyldimethylsilyl-4,6-bis-O-pyrenoyl-myo-inositol-1,3,5-orthoformate,32 and 1,8-diacrylnapthaline33 have been documented in literature. The unusual behavior of pyrene compounds has been explained due to formation π-stacking in the pyrene rings; such behaviour is known to be concentration dependent. With the increase in concentration, the pyrene rings approach close proximity to each other and dimerize due to π-stacking in an excited state. In other molecules with conformational flexibility, that too in presence of metal ions, metal ion-mediated conformational changes also result in two aromatic units close and undergo π–π stacking interaction either in ground state or excited state.34 In the present case of 9N3Me5Ox, due to conformational flexibility, the sterically congested 8HQ units are found to form π-stacking even in absence of metal ion. Molecular modeling studies of this molecule suggest that, in the ground state two 8HQ rings (out of three) in 9N3Me5Ox form parallel π-stacking, whereas the third ring displays perpendicular π-stacking with T-shaped geometry (Fig. 2(a)). Interestingly, the dimer of the compound in the excited state shows all six-rings π-stacking (Fig. 2(b)). Besides, in 9N3Me5Ox, three sets of proton donor (–OH) and proton acceptor (–N) groups are present in the same moiety; there is the possibility of excited state reaction in the molecule, particularly, the intramolecular proton transfer in an excited state (ESIPT).
 | ||
| Fig. 2 (a) 9N3Me5Ox DFT optimized structures, two 8HQ units are parallel stacked arranged and third ring perpendicular (T-shape) (b) intermolecular π–π stacking in dimer where all aromatic moieties are parallel stacked structure of 9N3Me5Ox. | ||
With these interesting findings, it is expected that any one of the factors or both the factors in cooperation may be responsible for the dual emission of the compound. To unfold the comprehensive picture of this unique behavior, an attempt has been made in this communication to investigate the detailed mechanism of dual fluorescence of 9N3Me5Ox by studying the ground state and excited state geometry, molecular modeling, 1H NMR, time-resolved fluorescence spectroscopic measurements, and also by the fluorescence spectroscopy of each species formed under varying concentration, pH and in solid form.
Result and discussion
The 8-hydroxyquinoline moiety has unique pyridyl N and OH structural characteristics. These N and OH pyridyl groups in the 8-hydroxyquinoline are able to act as a weak acid and a weak base under acidic and basic conditions, respectively, associated with the equilibrium between the protonated quinolinium NH–OH form and deprotonated quinoline N–O form;35 this moiety is able to act as the pH indicator for both acidic and basic systems. The protonation/deprotonation of the several acidic functions present in 8HQ could significantly affect the spectral properties of the molecule in ground (absorption) and excited (emission) state. 8-Hydroxyquinoline serves as a fluorophore because of its significant enhancement of fluorescence that occurs when the excited state intramolecular proton transfer (ESIPT) between the hydroxyl group and quinoline nitrogen is suppressed upon binding of a metal cation.33 However, some HQ derivatives are poorly luminescent34 due to an intramolecular photoinduced proton transfer (PPT) process between the hydroxyl group and the nearby quinoline nitrogen. In addition, in protic media, intermolecular PPT process involving solvent molecules can also occur, further decreasing the fluorescence quantum yield of this kind of fluorophore. The 8-hydroxyquinoline ligands are indeed characterized by strong intramolecular hydrogen bonding that may be photoinduced to undergo proton transfer in the excited state.36,37In the present tripod containing three units of 8-hydroxyquinoline, the acid–base behavior was studied through spectrophotometric and luminescence spectral studies in aqueous solution. Because of structural variation with pH, the studies were confined to three different regions: strongly acidic (pH ∼ 1.9): where the molecule exists in fully protonated form, strongly basic (pH ∼ 13): where the fully deprotonated form, and in neutral form at pH 7.5. These values were taken from a separate pH based titration experiment and calculated acid dissociation constants.
The electronic spectra of the three region are shown in Fig. 3. The concentration of the solution was maintained (1 × 10−5 M) to avoid any intermolecular interaction. The spectra show distinct features, which can be attributed to the existence of the different absorbing species in the ground state in cationic (protonated), neutral and anionic (deprotonated) forms. The absorption spectrum of fully protonated form under acidic conditions at pH ∼ 1.9 is characterized by two main absorption bands centered at 206 nm (ε = 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) and 250 nm (ε = 110000), and a very broad weak band at 330 nm (ε = 8000 M−1 cm−1) shown as in a Fig. 4. The first two bands are assigned to π–π* transitions whereas the broad band at 330 nm is assigned as n → π* transition.
000 M−1 cm−1) and 250 nm (ε = 110000), and a very broad weak band at 330 nm (ε = 8000 M−1 cm−1) shown as in a Fig. 4. The first two bands are assigned to π–π* transitions whereas the broad band at 330 nm is assigned as n → π* transition.
 | ||
| Fig. 3 Electronic spectra of 9N3Me5Ox (1 × 10−5 M) as a function of pH: (a) pH ∼ 1.9; (b) pH ∼ 7.5; (c) pH ∼ 10.9. Solvent: H2O, I = 0.1 M (KCl), T = 25.0 °C. | ||
 | ||
| Fig. 4 Fluorescence spectra of 9N3Me5Ox (1 × 10−5 M) in water (a) physiological pH ∼ 7.5, (b) pH ∼ 1.9, and (c) pH ∼ 10.9. Solvent: H2O, I = 0.1 M (KCl), T = 25.0 °C, with an excitation wavelength of 345 nm. | ||
In neutral pH, the π–π* absorption bands at 206 nm and 250 nm almost remain unaffected whereas the n–π* band of 330 nm was red-shifted and appeared at 345 nm (as shown at ‘b’ Fig. 3) because of deprotonation of protonated tertiary amines of 9N3Me5Ox. The n → π* transition absorption bands were broader and red-shifted as compared similar type reported tripod ligand indicating the presence of π-stacking interactions in the neutral medium. In basic condition, the electronic spectra of 9N3Me5Ox also exhibited two bands. The higher energy band was red shifted (about 8 nm) to 345 nm and the low energy band 250 nm was hypochromically and hypsochromically shift at to 244 nm, (ε = 40000 M−1 cm−1) [Fig. 3(c)], which are due to deprotonation of neutral hydroxyl function of 9N3Me5Ox to form completely deprotonated anion. This variation was observed up to pH 10.81.
The fluorescence emission spectra of 9N3Me5Ox in acidic, basic and neutral medium were studied in aqueous solution and are presented in Fig. 4. The fluorescence emission spectra displayed unusual behaviour at neutral pH. When excited at 345 nm, a normal fluorescence band at ∼375 nm with the large stokes-shift (∼30 nm), and a lower energy emission band at ∼456 nm were observed (‘a’ of Fig. 4). The observed fluorescence maxima were found to be independent of the excitation wavelength. The excitation spectra of the compound was also taken in the identical condition and presented in Fig. 5. The excitation spectra remain unaltered even with change in emission of wavelengths (viz., 375 nm and 456 nm) indicating the existence of only single species in the ground state.
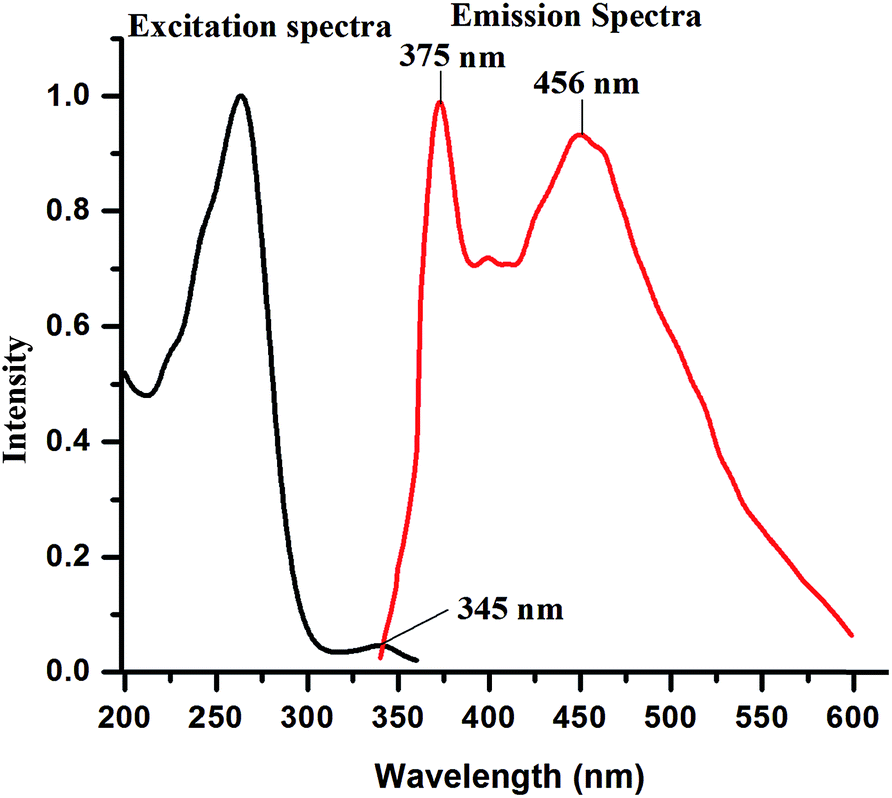 | ||
| Fig. 5 Excitation and emission spectra of 9N3Me5Ox (1 × 10−5 M) in water at pH ∼ 7.5 (λex = 345 nm). | ||
In acidic medium, when 9N3Me5Ox is excited at 345 nm wavelength, a fluorescence emission band centered at 375 nm was observed [Fig. 4(b)]. The second emission band which had appeared in neutral solution at 456 nm was disappeared. In basic medium, the second band was also disappeared; the only one emission band at 408 nm was observed [Fig. 4(c)].
A variety of organic compounds have been reported to show dual fluorescence in fluid solution, and the origin of which are so far assigned to one of the following mechanisms: (1) excited-state proton transfer or protonation/deprotonation,38 (2) intramolecular/intermolecular excimer/exciplex formation,39 (3) intramolecular electron transfer followed by conformational change and eventual intramolecular excimer formation in bridged electron donor–acceptor systems (the “harpooning” process)40 and (4) weak non-covalent intermolecular/intramolecular π–π stacking between aromatic rings which have conformational flexibility.41 In the light of the above mentioned possibilities and considering the interesting structural features of 9N3Me5Ox the observed dual fluorescence in aqueous neutral solution can be established with the following prospective.
Existence of intra-molecular π-stacking
Besides the conformational flexibility of pendant 8HQ units, the central cyclic 9N3 ring has an additional conformational flexibility. Due to rotation of several single bonds, the 8HQ pendant units become close or away from each other forming several conformations, out of which the π–π stacking interaction is possible in some form. A common feature of all π stacking is the close proximity of the reactants. The experimental onset of close proximity can be explored using 1H NMR spectroscopy. The 1H NMR experiment was performed for neutral 9N3Me5Ox to conform the existence of π stacking. DMSO-d6 was used for study neutral species, whereas DCl or NaOD was added to the neutral solution to study the spectra of acidic and basic species, respectively. The spectra are shown in Fig. 6. In the 1H NMR spectrum of 8HQ containing molecule well-resolved signals are expected characteristics to OH, CH, CH2 and CH3 protons. Signals due to CH of 8-hydroxyquinoline are always expected in the aromatic region in the range of 6.9–8.87 ppm which shows two doublets at 8.4 and 8.8 ppm, one double doublet at 7.5 and one triplet around 6.9 ppm. Singlets for one(–N–CH2) methylene proton of the cyclic ring and methylene (Ar-CH2) are expected at 2.0 and 3.4 ppm in 9N3Me5Ox. It can be seen from Fig. 6, signals of 8HQ in 1H NMR spectra of 9N3Me5Ox in acidic and basic medium were very well-resolved in comparison to that in the neutral medium. This difference in the spectra of three species is indicative for existence of π-stacking in the neutral solution of the molecule but not in acidic or basic condition. Since the structural architecture of the molecule 9N3Me5Ox is identical in acidic, neutral and basic medium, many conformers are also feasible for the molecule in acidic as well as basic medium due to conformational flexibility of 9N3 ring and rotation of single bonds which might be resulted by placing the three set of 8HQ rings close or away from each other. It is worth to mention that the repulsion of like positive charges in the protonated species and negative charges of phenolate ions in the deprotonated species hinder the quinoline rings to exist in close proximity, whereas no such repulsions exist in the neutral species (Fig. 7). This factor results close proximity of 8HQ pendant units only in the neutral species causing the π–π stacking in the molecule. Such type of π–π interaction is obviously absent in the 8HQ units in acidic and basic medium, due to repulsion of like charges of quinoline rings attached to the central unit of 9N3. Again, it is established that π-stacking interaction usually results in the shielding the protons in 1H NMR spectrum due to ring current effect of the aromatic rings.42,43 In the present case, due to existence of charges in protonated and deprotonated species the chemical shifts of the 8HQ ring protons were slightly more deshielded than the neutral condition (Fig. 6). | ||
| Fig. 6 Experimental 1H NMR spectra of 9N3Me5Ox, (a) acidic pH (b) basic (c) neutral pH. | ||
 | ||
| Fig. 7 9N3Me5Ox DFT optimized structures (a) protonated (b) neutral and (c) deprotonated. | ||
The spreading out of resonances signals due to shielding of different protons to different extents depend on their placement in the ring current effect have been documented by other molecules.44
In neutral condition, a board peak at 9.74 ppm for hydroxyl proton of quinoline ring was observed; this weak and broad observed band of hydroxyl protons is most probably resulted from intramolecular H-bonding of OH proton with N atom of quinoline unit.45
In addition to the evidences obtained from proton NMR, in silico studies were performed for various species. The conformational analysis of 9N3Me5Ox was done by molecular mechanics for protonated, deprotonated and neutral species using UFF force field. The schematic diagram of the derived lowest energy conformers of protonated, deprotonated and neutral species is shown in Fig. 7. No trace for π-stacking was noticed in the protonated or deprotonated species, whereas in neutral species, the π–π stacking interaction of three 8HQ was prominent. The three pendant rings on the macrocycle unit were not exactly parallel-superimposed. A parallel displaced type of π–π stacking between two rings was observed, whereas the third ring was perpendicular to theses two rings showing a perpendicular ‘T’ type of π stacking (Fig. 2(a)).
In order to confirm the energy profile of the π-stacking, a potential energy scan was performed at DFT level using B3LYP/631-G* method by studying the probable conformers of 9N3Me5Ox those might have resulted during rotation of single bonds attached of 8HQ rings attached to the 9N3 unit. The bonds were identified as D10, D21 and D36 (Fig. 8). Rotation was performed around the three dihedral D10, D21 and D36 angles separately in the macrocyclic ring in 18 steps with increment of 10° from 0–360° (Fig. 9). The results indicate that the geometry of the conformer in which three pendants are in π–π stacking arrangement has the lowest energy. Also, it was pragmatic that, the two 8HQ rings out of three in 9N3Me5Ox that were parallel to each other, showed parallel π-stacking, whereas the third ring displayed perpendicular π-stacking with T-shaped geometry (Fig. 9).
 | ||
| Fig. 8 Three dihedral angles viz., (a) D10, (b) D21 and (c) D36 that are rotate to obtain the conformers. | ||
 | ||
| Fig. 9 Potential energy scan about dihedral angle D10: (A), D21: (B), and D36: (C). | ||
On the basis of the evidences, viz., fluorescence spectra of solid sample and solutions, proton NMR and potential energy scan at DFT level, it could be established that there is occurrence of intramolecular π-stacking between the three 8HQ rings of the pendant macrocycle, wherein there is a parallel displaced π-stacking between two rings and one T-type (more appropriately π-type) π-stacking, perpendicular to the third [Fig. 2(a)].
Existence of intermolecular π-stacking and excimer formation
In principle, a single ground state species produces a single emission peak. But, in the present case, as mentioned earlier, two emission peaks were observed for the referenced molecule in the neutral condition. This observation implied the existence of two different species of the molecule in excited state. Majority of examples of compounds having rigid aromatic structures exhibiting dual fluorescence have been explained by formation of excimers, that too because of existence π-stacking interaction in the excited state. On the basis of the NMR experiments, solution fluorescence spectra and molecular modeling, the existence of intramolecular π-stacking has been established in the ground state. Excimer formation is concentration dependent; with increase in concentration of solution, the monomers dimerize to form excimer in the excited state. In order to ascertain the above probability, a concentration dependent emission spectra experiment was performed. The spectra were taken in varying concentration of the compound from 10−5 to 10−9 M at 345 nm excitation wavelength (Fig. 10). At low concentration, i.e., at 10−9 M, a single emission band at 375 nm was observed; this is assigned to be the monomer band. With the gradual increase in the concentration of the ligand from 10−9 to 10−5 M, the intensity of monomer band at 375 nm decreased progressively with simultaneous appearance of an additional red-shifted non-structured emission band, centered at 456 nm. The new band at 456 nm may be assigned to the excimer emission. The complete disappearance of monomer peak was observed at very high concentration, 5.0 × 10−2 M, indicating that all molecules form excimers at that concentration (Fig. 10). | ||
| Fig. 10 Effect of the concentration on fluorescence spectra of 9N3Me5Ox in water (a) 5.0 × 10−2 M, (b) 1.0 × 10−5 M, (c) 1.0 × 10−6 M, (d) 1.0 × 10−7 M, (e) 1.0 × 10−8 M, (f) 1.0 × 10−9 M, (g) solid state (black line) emission spectra. λex = 345 nm. | ||
In order to testify the excimer formation, a molecular modeling calculation was performed using molecular mechanics taking two DFT optimized monomers. Interestingly, the lowest energy structure resulted a dimer with all the rings showing π-stacking. The molecule with three pairs parallel π-stacking structure containing all six rings is an unique of its kind exhibiting intramolecular as well as the intermolecular π-stacking as illustrated in Fig. 2(b).
8HQ is a bifunctional hydrogen-bonding molecule that simultaneously acts as a H-donor at the OH site and an acceptor at the N-atom in aqueous or alcohol solution. Upon photoexcitation, the acid/base properties of this molecule change significantly at both the sites, rendering OH-group more acidic and the N-atom more basic. Also, the acidic (H-bond donating) and basic (H-bond accepting) groups of the molecule are close each other and hence a single solvent can bind to both sites simultaneously and monomer molecules can arrange to form dimer via H-bonding (Fig. 11). One might expect tautomerization in H-bond accepting via intra-molecular hydrogen bonding and in all other solvent solutions, there may exist a competition between intra and intermolecular H-bonding.46 To explain the fluorescence behavior of above possible species, the steady state absorption, fluorescence excitation and emission spectra of 8-hydroxyquinoline have been studied in wide range of solvents of varying dipole moments, concentrations and excitation wavelengths. The finding of the studies revealed that the exhibition of dual fluorescence is possible due to the formation of the stoichiometric46 1:1 and 1:2 hydrogen bonding complexes in the ground state as well as excited state. Recently, Park et al.,47 have investigated the origin of ultra weak fluorescence of unsubstituted 8HQ in aqueous solution in acidic, neutral and basic condition but did not notice any existence of dual fluorescence. However, they have concluded that 8HQ, as well as its protonated or deprotonated forms, undergo ultrafast excited state proton transfer within femto- to picoseconds to produce a tautomer as product, the lifetime of which is also ultrashort. Through molecular dynamics they showed that, 8HQ in water mainly exists as non-cyclically H-bonded to adjacent water molecules with the two acidic enol and the basic imine functional groups being independently hydrated. Relatively small fraction of 8HQ form cyclically H-bonded complex with a water molecule. The non-cyclically H-bonded 8HQ is proposed to undergo photoinduced prototropic tautomerization stepwise via forming the anionic species as a reaction intermediate to yield a tautomer as a product in the excited state.
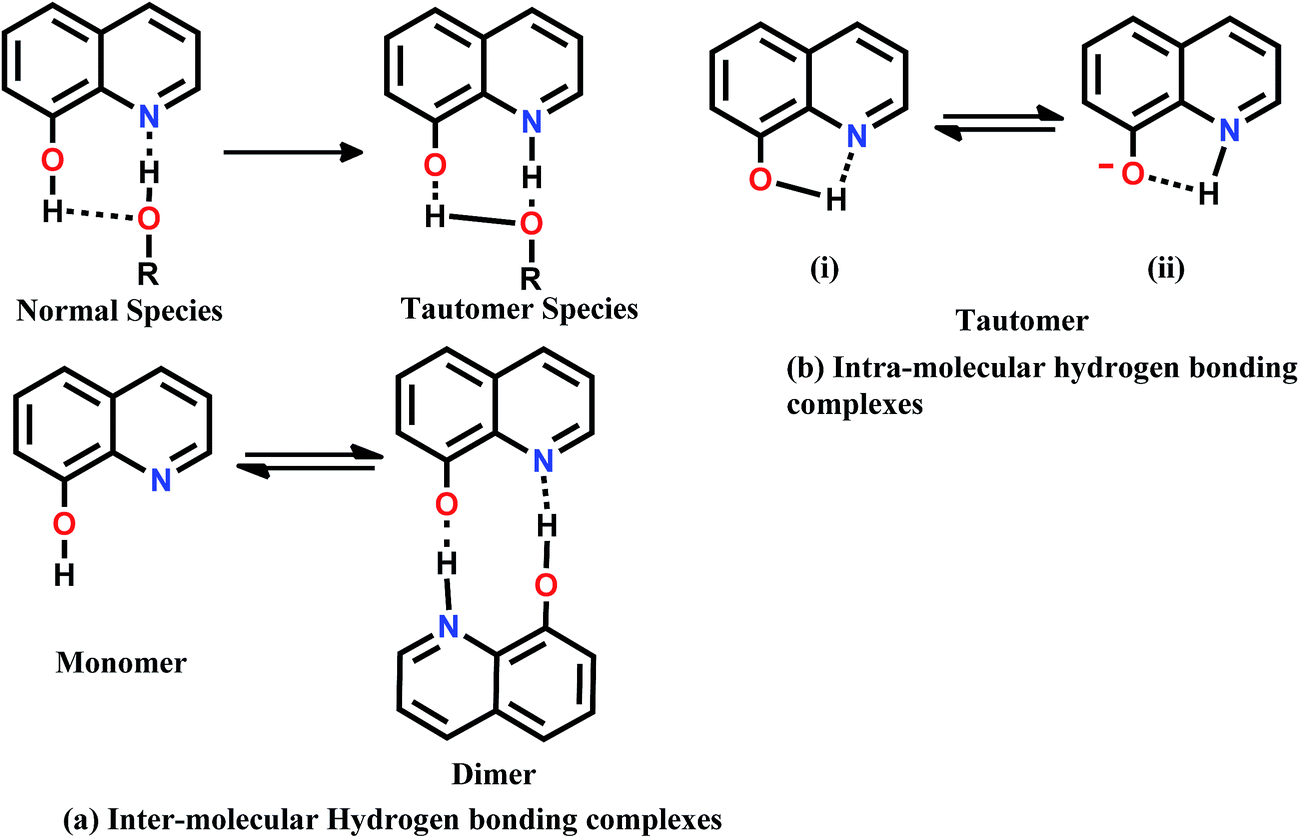 | ||
| Fig. 11 Possible resonance structures of 8-hydroxyquinoline. | ||
The proton-transfer dynamics is found to be more facile compared to that of other hydroxyquinolines, resulting in the ultrashort lifetimes of all the involved excited species: 3 ps for the parent 8HQ; 700 fs for the anionic intermediate; and 10 ps for the tautomer as a product. This finding was the main reason for very weak fluorescence of 8HQ in aqueous solutions in contrast to well-known strong fluorescence of 8HQ when chelated with metal ions without undergoing proton transfer. When 8HQ binds to metal cations, the ligand is on the deprotonated form, which cannot undergo the ultrafast proton transfer without generating the short lived dark product, but survives much longer to tens of nanosecond to “turn on” fluorescence. In order to ascertain any possibility of prototropic involvement in 9N3Me5Ox, fluorescence emission spectra were obtained in polar protic, polar aprotic and non polar solvents at the same experimental conditions. Prominent dual fluorescence was observed in water, ethanol, methanol and acetonitrile (Fig. 12), whereas practically only one band was observed in non-protic solvent like dichloromethane and dimethylsulfoxide. These results indicate the involvement and assistance of proton or prototropic reactions in the excited state in the exhibition of dual fluorescence.
 | ||
| Fig. 12 Fluorescence emission spectra in different solvent at 1.0 × 10−5 M at 25 °C (a) H2O, (b) acetonitrile (ACN), (c) ethanol (EtOH), (d) dichloromethane (DCM), (e) dimethylsulfoxide (DMSO) and (f) methanol (MeOH). | ||
In order to strengthen the urging factors of dual fluorescence in 9N3Me5Ox in the aqueous medium at the neutral solution, time-resolved fluorescence spectroscopy study was carried out by measuring decay constants for specific species present in the excited state using the nanosecond time-correlated single photon counting (TCSPC) technique. The experiments were also performed in acidic and basic pH for comparison. In time-resolved experiments care was taken to avoid scattered light, therefore experimental artifacts cannot be considered for the shorter component. Fluorescence decays were recorded for two emission profiles, one at 375 nm and the other at 456 nm with λex = 345 nm. The fluorescence decay curves are shown in Fig. 13 and the decay data are summarized in Table 1. Fig. 13(a) shows the time-resolved fluorescence spectra of the aqueous solution of 9N3Me5Ox at neutral pH ∼ 7.5 whereas the spectra of acidic and basic solutions are presented in (b) and (c) of Fig. 13. All these fluorescence curves were best fitted with the biexponential decay function and the time decays calculated. The τ values were found to be 0.27 ns and 0.20 ns for 375 nm and 456 nm emission respectively indicating existence of two different species in the neutral state. It is known that the conversion of enol to keto form through ESIPT is a slow process and exhibit higher value of decay constant as compared to the individual form.27 The observed shorter decay time of ∼0.20 ns of the molecule can be attributed to non-existence of keto–enol tautomerization in excited state thereby ruling out the occurrence of any ESIPT process in the molecule. Conversely, the short-lived species causing the additional band in the fluorescence spectra of neutral solution is due to formation of intermolecular π-stacked excimer caused by excited reaction of the intramolecular π-stacked monomers.
| λex (nm) | λem (nm) | τ1a (ns) | τ2a (ns) | β1c (ns) | β2c (ns) |
|
χ2b |
|---|---|---|---|---|---|---|---|
| a Decay time (τ) is in ns. Standard deviation is given in parentheses.b χ2 is the chi-square value for best fit either for single exponential fit or for double exponential fit.c β’s are relative amplitudes. | |||||||
| 345 | 375 (pH 1) | 2.71 (0.066) | 0.146 (0.014) | 0.3404 | 0.0718 | 0.93 | 1.195 |
| 375 (pH 7) | 0.857 (0.019) | 6.17 (0.031) | 0.2691 | 0.007 | 0.27 | 1.26 | |
| 456 (pH 7) | 0.407 (0.042) | 4.07 (0.027) | 0.338 | 0.017 | 0.20 | 1.26 | |
| 408 (pH 14) | 0.916 (0.048) | 2.94 (0.017) | 0.087 | 0.089 | 0.34 | 1.29 | |
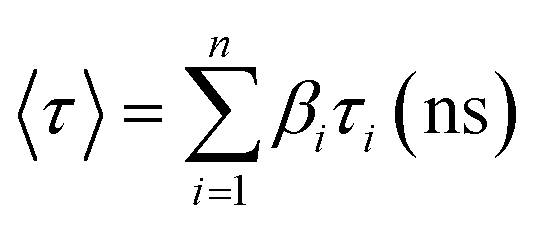
 | ||
| Fig. 13 Fluorescence decay curve of 9N3Me5Ox in water at pH ∼ 7.5 (a), pH ∼ 1.9 (b) and pH ∼ 10.9 (c). | ||
Conclusions
The excimer with three pairs of parallel π-stacking structures containing all six 8HQ rings is unique of its own kind exhibiting intramolecular as well as the intermolecular π-stacking. Existence of intramolecular π-stacking interaction in ground state, predicted by the molecular modeling was confirmed by 1H NMR spectroscopic evidences. Variable concentration fluorescence spectroscopic studies spot out the intermolecular π–π stacking between the two molecules of 9N3Me5Ox in excited state. At low concentration, only single monomer band existed, whereas, with increase in concentration of the solution, the intensity of the monomer band drops off with appearance of a new band causing dual fluorescence resulted from excited state dimerization of 9N3Me5Ox through intermolecular π–π stacking. At high concentration, as well as in the solid state fluorescence spectrum, only one band was appeared due to excimer emission. The possibility of any excited state proton transfer in the molecule is ruled out from the time decay measurement. In addition, the assistance of polar protic solvent in π–π stacking interaction in the excited state was established by the variable solvent fluorescence spectral report. With substitution of more 8HQ units to macrocycle 9N3, the life time of the resulted molecule was enhanced to nanosecond scale, as compared to the reported short-lived unsubstituted 8HQ which has lifetime in picoseconds level, thus tuning “fluorescence on”.47 Although the existence of dual fluorescence of 9N3Me5Ox could be established on the basis of interpretation of experimental results, the detailed quantum mechanical calculations for interpretation of the observed phenomena is in progress.Acknowledgements
One of the authors (Rohini) is grateful to the Sant Longowal Insititute of Engineering and Technology, Longowal, (SLIET) for award of institute research fellowship. Support of Professor A. K. Mishra (IIT, Madras) for his valuable suggestions and help in lifetime measurement is gratefully acknowledged. The authors also thank to Dr K. K Haldar for helpful discussions.References
- J. J. Fox, J. Chem. Soc., 1910, 97, 1119 RSC
.
- S. F. Mason, J. Philp and B. E. Smith, J. Chem. Soc. A, 1968, 3051 RSC
- Z. Yin, B. Wang, G. Chen and M. Zhan, J. Mater. Sci., 2011, 46, 2397 CrossRef CAS
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS
- M. Colle, R. E. Dinnebier and W. Brutting, Chem. Commun., 2002, 23, 2908 RSC
- L. G. C. Rego, R. da Silva, J. A. Freire, R. C. Snoeberger and V. S. Batista, J. Phys. Chem. C, 2010, 114, 1317 CAS
- L. Duan, L. Wang, F. Li, F. Li and L. Sun, Acc. Chem. Res., 2015, 48, 2084 CrossRef CAS PubMed
- M. Albrecht, M. Fiege and O. Osetska, Coord. Chem. Rev., 2008, 252, 812 CrossRef CAS
- S. Cho, M. W. Mara, X. Wang, J. V. Lockard, A. A. Rachford, F. N. Castellano and L. X. Chen, J. Phys. Chem. A, 2011, 115, 3990 CrossRef CAS PubMed
- M. A. Palacios, Z. Wang, V. A. Montes, G. V. Zyryanov and P. Anzenbacher Jr, J. Am. Chem. Soc., 2008, 130, 10307 CrossRef CAS PubMed
- H. Zhang, L.-F. Han, K. A. Zachariasse and Y.-B. Jiang, Org. Lett., 2005, 7, 4217 CrossRef CAS PubMed
- N. M. Shavaleev, R. Scopelliti, F. Gumy and J.-C. G. Bunzli, Inorg. Chem., 2009, 48, 2908 CrossRef CAS PubMed
- L. L. Merritt Jr and J. K. Walker, Ind. Eng. Chem., 1944, 16, 387 Search PubMed
- G. P. Demopoulos and P. A. Distin, Hydrometallurgy, 1983, 11, 389 CrossRef CAS
- A. M. Mancino, S. S. Hindo, A. Kochi and M. H. Lim, Inorg. Chem., 2009, 48, 9596 CrossRef CAS PubMed
- J.-S. Choi, J. J. Braymer, R. P. R. Nanga, A. Ramamoorthy and M. H. Lim, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21990 CrossRef CAS PubMed
- P. J. Crouch and K. J. Barnham, Acc. Chem. Res., 2012, 45, 1604 CrossRef CAS PubMed
- S. H. Chan, C. H. Chui, S. W. Chan, S. H. Kok, D. Chan, M. Y. Tsoi, P. H. Leung, A. K. Lam, A. S. Chan, K. H. Lam and J. C. Tang, ACS Med. Chem. Lett., 2012, 4, 170 CrossRef PubMed
- K. Mekouar, J.-F. Mouscadet, D. Desmaele, F. Subra, H. Leh, D. Savoure, C. Auclair and J. d'Angelo, J. Med. Chem., 1998, 41, 2846 CrossRef CAS PubMed
- Y. Chen, H. Wang, L. Wan, Y. Bian and J. Jiang, J. Org. Chem., 2011, 76, 3774–3781 CrossRef CAS PubMed
- F. Launay, V. Alain, E. Destandau, N. Ramos, E. Bardez, P. Baret and J. L. Pierre, New J. Chem., 2001, 25, 1269–1280 RSC
- L. P. Jones, J. A. Amoroso, C. J. Jeffery, A. J. McCleverty, E. Psillakis, H. L. Rees and D. M. Ward, Inorg. Chem., 1997, 36, 10–18 CrossRef
- R. Akbar, M. Baral and B. K. Kanungo, RSC Adv., 2015, 5, 16207–16222 RSC
- R. Akbar, M. Baral and B. K. Kanungo, J. Chem. Eng. Data, 2015, 60, 3236–3245 CrossRef CAS
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum, New York, 3rd edn, 1983, ch. 1, pp. 2–5 Search PubMed
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum, New York, 3rd edn, 1983, ch. 1, p. 9 Search PubMed
- G. Venkataramana and S. Sankararaman, Org. Lett., 2006, 8, 2739–2742 CrossRef CAS PubMed
- S. Sankararaman, G. Venkataramana and B. Varghese, J. Org. Chem., 2008, 73, 2404–2407 CrossRef CAS PubMed
- R. Nandy, M. Subramoni, B. Varghese and S. Sankararaman, J. Org. Chem., 2007, 72, 938–944 CrossRef CAS PubMed
- N. K. Joshi, P. Arora, S. Pant and H. C. Joshi, Photochem. Photobiol. Sci., 2014, 13, 929 CAS
- A. Pramanik and G. Das, Tetrahedron, 2009, 65, 2196–2200 CrossRef CAS
- M. Kadirvel, B. Arsic, S. Freeman and E. V Bichenkova, Org. Biomol. Chem., 2008, 6, 1966–1972 CAS
- X. Mei and C. Wolf, J. Org. Chem., 2005, 70, 2299–2305 CrossRef CAS PubMed
-
(a) H. Yuasa, N. Miyagawa, T. Izumi, M. Nakatani and M. Izumi, Org. Lett., 2004, 6, 1489–1492 CrossRef CAS PubMed
- Y. Mei, P. A. Bentley and W. Wang, Tetrahedron Lett., 2006, 47, 2447–2449 CrossRef CAS
- G. K. Vaswani and D. K. Mark, Inorg. Chem., 2009, 48, 5797–5800 CrossRef PubMed
- V. Guallar, M. Moreno, J. M. Lluch, F. Amat-Guerri and A. Douhal, J. Phys. Chem., 1996, 100, 19789–19794 CrossRef CAS
- M. H. Van Benthem and G. D. Gillispie, J. Phys. Chem., 1984, 88, 2954 CrossRef CAS
- T. Kumpulainen and A. M. Brouwer, Chem. Phys., 2012, 14, 13019–13026 CAS
- D. W. Cho, M. Fujitsuka, K. H. Choi, M. J. Park, U. C. Yoon and T. Majima, J. Phys. Chem. B, 2006, 110, 4576–4582 CrossRef CAS PubMed
- J. W. Verhoeven, Pure Appl. Chem., 1990, 62, 1585–1596 CrossRef CAS
-
(a) A. Fechtenkotter, K. Saalwachter, M. A. Harbison, K. Mullen and H. W. Spiess, Angew. Chem., Int. Ed., 1999, 38, 3039–3042 CrossRef CAS
- K. Katagiri, T. Tohaya, H. Masu, M. Tominaga and I. Azumaya, J. Org. Chem., 2009, 74, 2804–2810 CrossRef CAS PubMed
- R. Nandy, M. Subramoni, B. Varghese and S. Sankararaman, J. Org. Chem., 2007, 72, 938–944 CrossRef CAS PubMed
- M. Albrechet, K. Witt, R. Frohlich and O. Kataeva, Tetrahedron, 2002, 58, 561–567 CrossRef
- L. R. Naik and N. N. Math, Indian J. Pure Appl. Phys., 2005, 43, 743 CAS
- S. Y. Park, P. Ghosh, S. O. Park, Y. M. Lee, S. K. Kwak and O.-H. Kwon, RSC Adv., 2016, 6, 9812–9821 RSC
| This journal is © The Royal Society of Chemistry 2016 |