
Study on Fe–Co alloy role over RANEY® Fe–Co bimetallic catalysts in Fischer–Tropsch synthesis
Abstract
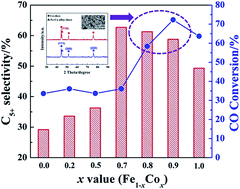
To further understand the role of Fe–Co alloys in Fischer–Tropsch synthesis, a series of RANEY® Fe–Co mono/bimetallic catalysts were prepared through alkali leaching solidified Fe–Co–Al alloy. There are three advantages about these catalysts: (i) RANEY® structure can provide certain surface area for catalytic reaction; (ii) these catalysts avoid the influence of the metal–support interaction due to the absence of support; (iii) the composition of active phase can be determined easily. The FT results showed that not all the Fe–Co alloy could enhance the C5+ selectivity, only when the ratio of Fe/Co is 3 : 7 can C5+ selectivity be promoted effectively. Besides, we also found that Fe–Co alloy did not show intended synergy effects on the FT activity, instead, Co-dominant catalysts exhibited a higher CO conversion.
 Please wait while we load your content...
Please wait while we load your content...

