DOI:
10.1039/C6RA22721J
(Paper)
RSC Adv., 2016,
6, 98128-98140
High-performance electromagnetic wave absorbing composites prepared by one-step transformation of Fe3+ mediated egg-box structure of seaweed†
Received
12th September 2016
, Accepted 10th October 2016
First published on 10th October 2016
Abstract
Composites incorporating ferromagnetic components into a highly porous carbon matrix are promising as electromagnetic wave absorption materials; however, the design of a facile preparation process with scale-up potentiality still remains a practical challenge. Herein, a new kind of three-dimensional (3D) magnetic carbonaceous bead-like (MCB) Fe–SA-X composites has been directly prepared using a Fe3+ mediated egg-box structure of renewable sodium alginate as single starting material and a controllable carbonization process; as compared to previous work on carbon-based microwave absorbers, it avoids tedious and time-consuming preparation procedures. It was verified that, according to various characterization results, the atmosphere and temperature for the carbonization of Fe–SA precursors were crucial factors for the formation of the ferromagnetic metal particles and carbon matrix in the porous Fe–SA-X composites. Among the Fe–SA-X composites obtained at different temperatures, Fe–SA-600 obtained at 600 °C exhibited the best performance for electromagnetic wave absorption, for which can be attributed to the synergy of magnetism of Fe3O4 nanoparticles and the nano-network of graphitized carbon. The maximum reflection loss (RL) of Fe–SA-600 reached −24 dB, and the effective absorption bandwidth (RL ≤ −10 dB) was 4.80 GHz (13.2–18 GHz) corresponding to an absorber thickness of only 1.5 mm. Such excellent electromagnetic absorption properties could be assigned to the improvement of the impedance match and interfacial polarization and unique porous structures, by which can lead to the effective microwave multi-reflection and scattering. This kind of composite is an attractive candidate for new types of high-performance electromagnetic wave-absorbing materials, meeting for the current requirements of wide-band absorption, high efficiency absorption capability, thin thickness and light weight.
1. Introduction
As is well known, to solve the electro-magnetic interface (EMI) problem, developing high-performance microwave absorption materials is necessary,1,2 that is to say, converting the incidence electromagnetic wave into thermal energy and refraining from reflection on the surface of the absorber.3 In a general way, the high-performance absorbers need to fill the bill for low reflection loss values RLmin, broad effective frequency (fE), and lower density and good thermal stability characteristics simultaneously.4 The expected reflection loss value must be less than −10 dB. As a matter of fact, it is not difficult to obtain great reflection loss values, e.g. MnO2 nanorods (−25 dB), Ni fiber (−39.5 dB), dendritic-like Fe (−32.3 dB).5–7 However, inadequate frequency width (fE), large coating thickness (t) and high density may confine their further potential applications. Generally speaking, a broad fE value, thin coating thickness (less than 2 mm) and lower density are essential in our real life.
Naturally, only doing meaningful research studies, one would obtain such kind of absorbers. For instance, several materials such as metals, ferrites, conducting polymers and carbons, for which with advantages and disadvantages when applied alone, can be applied for electromagnetic wave absorption.8–10 Another example, the frequency ranges of ferromagnetic metals such as Fe, Ni and Co their alloys are always narrow, at the same time exhibit strong absorption intensity.11 Comparatively speaking, depending on high dielectric constant, low density, large surface area and excellent thermal stability, carbon-based materials such as CNTs and graphene, carbon nanofibers (CNFs), carbon nanorods, carbon nanospheres, carbon nanowires, can be good lightweight dielectric absorbing.12,13 In addition, carbon and iron are nontoxic, low-cost and abundant, simultaneously. Meanwhile, carbon-based and iron-based composite materials with excellent electromagnetic absorption properties can fill the bill of electromagnetic wave absorption materials.14 Therefore, those composites of iron and carbon species may have been a promising candidate for lightweight microwave absorption materials; and a number of studies have been done on the kind of composites for microwave shielding. For example, compared with the pure carbon nanomaterials, Fe3O4@Ppy@CNT, and Fe3O4@SiO2@RGO, r-GO@Fe3O4 (ref. 15–17) may have a important increased impedance matching because of the reduced dielectric constant. Nevertheless, although these composites are provided with expected absorbing characteristics, the high-weight, high-cost, low-yield, and more important complicated preparation processes, are restrictive factors toward their practical applications.
In view of these issues, researchers began to shift from conventional ideas to a new direction that converses renewable biomass into high-performance microwave absorbers. For instances, as abundant renewable resources, biomasses were paid increasing attention in the preparation of potential carbonaceous functional materials.18–20 Particularly, intensive attention has been paid to the utilization of bountiful sea resources.21 Among the natural polymers that with abundant carboxyl and hydroxyl groups in its polymeric matrix,22 which possess advantages example high molecular-weight, bio-gradation and low-cost sodium alginate (SA) and so on. What's more, alginate macromolecules can be cross-linked with metal ions, such as Fe3+, Co2+ and Ni2+, etc. The complex structures can be represented by the alleged “egg-box” model, in which each metal ion is harmonious to the carboxylate and hydroxyl groups of four G monomers from two interlink chains of alginate macromolecules.23,24 Such a significant structural characteristic of metal–alginate complexes may provide a new method for the synthesis of novel 3D carbonaceous nano-materials with mature nano-network and scattered metal (metal oxides) particles by employing controllable carbonization process in an inert atmosphere, considering the truth that the carbonization of the M–alginates might form metal (oxide) NPs encapsulated by protective carbon layers.22 However, in contrast to reported extensive studies on electrodes, sorbents and catalytic materials, the exploration of another potential application of carbon encapsulated metal (oxide) NPs prepared using unique metal-meditated “egg-box” structure of polymeric alginate as effective electromagnetic absorbing materials has rarely been reported.
To demonstrate such a concept, herein we report a simple and effective biomass conversion strategy for a new class of porous graphitic carbon composites with excellent microwave absorbing performance. As for the preparation of the targeted composites, the coordination with four α-L-gu-luronate blocks of alginate is used to immobilize Fe3+ cations into novel “egg-box” for making seaweed hybrid aerogels.25–28 We should emphasize the merits of our synthetic approach again, compared to conventional electromagnetic wave absorbers, this material is more advantageous. Most of the conventional electromagnetic wave absorbers in the choice of dielectric materials often need to modify the dielectric material and pretreatment to improve the internal structure. Although some good electromagnetic wave absorption performance can be achieved in this process, complicated preparation routes, high cost and environmental pollution cannot be ignored. The introduction of biomass alginate greatly reduced these shortcomings. Biomass, as a bountiful renewable nontoxic resource, is more attractive in the preparation of useful carbon materials. The alginate is cross-linked with Fe3+ to form a gel, by controlling the carbonation temperature to indirectly control the magnetic media composition and content, to improve the absorption properties. The synthesis is a really facile, direct, environmentally friendly, and controllable pathway due to the seaweed precursor is sustainable and earth-abundant. It is highly potential to scale-up the synthesis into an industrial level since the method is very simple and cost-effective. More importantly, it can also achieve better electromagnetic absorption properties. As for its unique pore structure, which greatly increased the incident electromagnetic waves in the material within the reflection and scattering, coupled with the mutual interaction of magnetic media, this weakening effect is even more obvious.
2. Experimental section
2.1. Materials
Sodium alginate and FeCl3·6H2O were purchased from Beijing XinDingTengFei Co. Ethanol was purchased from Sinopharm Chemical Reagent Factory, China. All chemicals were of analytical grade and used as received without any further purification. Distilled water was used in all experiment process.
2.2. Preparation of Fe–SA beads
Two gram of sodium alginate was added to 100 mL distilled water and stirred vigorously for 12 h. Then the above mixture was added dropwise into 5% FeCl3 (w/v) solution for cross-linking to get SA–Fe(III) hydrogel beads and left undisturbed in Fe(III) solution for 6 h for complete cross linking for obtaining iron–alginate hydrogel beads. Finally, the beads were washed with distilled water followed by ethanol to remove extra Cl− and Fe3+ ions. Then, the as-prepared hydrogels beads were transferred to a freezer at −50 °C for 12 h and dehydrated via a freeze-drying process in a vacuum freeze dryer for 24 h to obtain dried SA–Fe(III) beads.
2.3. Preparation of the MCB Fe–SA-X
The dried SA–Fe(III) beads was placed in a tube furnace and heated at a rate of 5 °C min−1 to a setted up temperature (600, 700, 800 or 900 °C) in N2 atmosphere and kept for 2 h. After being cooled in flowing N2, the obtained materials were recorded as Fe–SA-X (X = 600, 700, 800 or 900 °C), corresponding to employed heating temperature.
2.4. Materials characterization
The morphology of the samples was investigated by field-emission scanning electron microscope (SEM, JEM JEOL 2100) and transmission electron microcopy (TEM, Hitachi H9000NAR), respectively. The composition and phase purity of the as-prepared sample were analyzed by X-ray diffraction, X-ray diffraction (XRD) patterns were obtained with a Rigaku model D/max-2500 diffractometer with Cu-Kα radiation (λ = 1.540 Å) at 40 kV, 10–80° at 8° min−1 scanning speed. The chemical bonds were analysed using a KBr pellet technique by Fourier transform infrared spectroscopy (FT-IR, Perkin-Elmer, USA). The specific surface area and pore diameter of the samples were performed by nitrogen adsorption–desorption experiments (Quantachrome AsiQM0000-3, USA). X-ray photoelectron spectroscopy (XPS) measurements were performed using the he PHI 5000 VersaProbe systems. The Raman spectra of the composites were measured using a Renishaw in Via Raman Microscope. The magnetic properties of the as-prepared Fe–SA-X composites were measured by (VSM, Lake Shore 7400) at room temperature.
2.5. Electromagnetic wave absorption measurements
For composite sample used for EM absorption measurement was prepared by mixing Fe–SA-X nanocomposites with paraffin. The content of paraffin wax was debugged and finally fixed to 25 wt%. The mixtures were then pressed into cylindrical-shaped samples with Φin of 3.04 mm, Φout of 7.00 mm and thickness of 2.00 mm. The relative permittivity and relative permeability values were measured in the 2–18 GHz range with the coaxial line method by an Agilent N5222A vector network.29 The reflection loss (RL) was calculated according to the following equations:| |
Zin = Z0(μr/εr)1/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) tanh[j(2πfd)/c(μrεr)1/2] tanh[j(2πfd)/c(μrεr)1/2]
| (1) |
| | |
RL = 20log|(Zin − Z0)/(Zin + Z0)|
| (2) |
where f is the frequency, d is the thickness of absorber, c is the velocity of electromagnetic waves in free space, μr(μr = μ′ − jμ′′) and εr(εr = ε′ − jε′′) are the relative complex permeability and permittivity, respectively, Z0 stands for the impedance of free space and Zin means the input impedance of the absorber.
3. Results and discussion
3.1. Characterization of Fe–SA-X
The manufacturing process of Fe–SA-X materials was mentioned in Scheme 1. First, via the electronic attraction, the carboxyl group of SA can form a complex with Fe(III). The results of the process is that the binding of numerous iron ions by ion exchange between sodium and ferric ions. Next step is to obtain SA–Fe(III) beads by the process in a vacuum freeze dryer for 24 h. The obtained SA–Fe(III) beads, at diverse carbonization temperature under an N2 atmosphere, were treated and the carbonaceous samples can be researched for electromagnetic wave absorption performance. The fresh way is scalable to meet different industrial production levels and sufficiently versatile to incorporate a wide range of metals for various metal/carbon or metallic oxide/carbon composites.
 |
| | Scheme 1 The fabrication process of seaweed-derived electromagnetic wave absorbing composites using Fe3+ gelled sodium alginate as a single starting material. | |
Firstly, SEM was used to study the detailed microscopic structures and morphologies of the Fe–SA-X prepared at different temperatures, and the representative images are shown in Fig. 1. As we can see, when the carbonization temperature reached 600 °C, it can be observed that a large amount of nanoparticles uniformly distributed on the surface of the materials, with an average particle size of about 25–30 nm without aggregation; this mainly is associated with the protective nano-network of carbon due to the shrinkage of alginate polymer during the carbonization progress.30 With the increase of the carbonization temperature, the pore structure destroyed significantly and the particle size increased. Possibly, this transformation was favorable to the formation of carbonaceous composites with enhancing surface area, and it can be confirmed by the following BET analysis. Moreover, for Fe–SA-600, the high porosity and highly dispersed metal (oxides) nanoparticles might be favorable for the absorption of electromagnetic waves.31
 |
| | Fig. 1 SEM images of (a) Fe–SA-600, (b) Fe–SA-700, (c) Fe–SA-900. | |
In order to investigate the distribution of metal-based particles in the carbon materials, the TEM results of the different carbonization temperature samples were conducted. The images of the samples were depicted in the Fig. 2. It can be observed that a large amount of metal (or oxides) nanoparticles uniformly distribute throughout the carbon network, with an average particle size of about 20 nm to 25 nm (see inset of Fig. 2a). The high degree of distribution of magnetic particles is mainly due to the electrostatic attraction between Fe3+ ions and –COOH groups in the sodium alginate.32 Meanwhile, some tubulose structures were found in the TEM micrograph (see inset of Fig. 2b); and the mean diameter of tubes was 25 nm. With increasing the carbonization temperature, the particle size increased gradually. When the carbonization temperature reached 900 °C, the magnetic particles were finely wrapped by the graphene foam which derived from the carbonization of alginate polymer (see inset of Fig. 2c).33 The loading of magnetic nanoparticles on the foam benefited to increase the specific surface area and render the foam with magnetic property. Meanwhile, the increased particle size obstructs the formation of good EM wave passages; it would be detrimental to their further transmission within the material. Possibly, it may be not conducive to the absorption of electromagnetic waves.34
 |
| | Fig. 2 TEM images of (a), (b) for Fe-SA-600, and (c) for Fe-SA-900. | |
The structures and phase of the synthesized composites were characterized by XRD. As shown in Fig. 3, the peaks at 2θ = 26.2° in Fe–SA-X (X = 600, 700, 800, 900) which can be assigned to the typical graphitic (002) plane of the graphite structure,35 were due to the carbonization of organic alginate polymer under high temperature, resulting in carbon structures with some degree of graphite order.38 In addition, the XRD patterns of Fe–SA-X (X = 600, 700, 800, 900) composites (upper) were obtained from data bank entry (lower). For Fe–SA-600, the characteristic diffraction peaks at around 2θ = 35.6°, 43.3° and 62.8° are corresponding to the (311), (400) and (440) planes of Fe3O4 (JCPDS, no. 89-0688). The other diffraction peaks at around 2θ = 45.1° and 65.166° can correspond to the (031) planes of Fe3C (JCPDS, no. 41-1487) and Fe,36 respectively. With the increase of the carbonization temperature, the peaks for Fe3C increased, while the peaks of Fe3O4 all decreased. To our best acknowledge, the decreasing of Fe3O4, a typical magnetic loss material for EM wave absorption, was unfavorable to EM wave absorption.32 When the carbonization temperature reached 900 °C, those peaks observed in the Fig. 3 can be attributed to the graphite, Fe3C, Fe and Fe3O4, respectively. Additionally, it can be noted that the amount of Fe3C increased significantly, while Fe3O4 decreased.37 Therefore, the ingredient of this carbon–iron composite material can be easily changed by carbonizing Fe(III)-mediated egg-box structures. It also disclosed that the amount of Fe3O4 nanoparticles decreased whereas the carbonization temperatures increasing; this verdict is further sustained by the following FTIR results.
 |
| | Fig. 3 XRD of Fe–SA-600, Fe–SA-700, Fe–SA-800, Fe–SA-900. | |
In addition, Fig. 4 shows a comparison of the FT-IR spectra of Fe–SA-600 and Fe–SA-800. It can be observed from the FTIR spectrum of the Fe–SA-600 that a band appeared at around 3440 and 1626 cm−1 can be assigned to O–H and C–O stretching vibrations, respectively; implying the presence of numerous hydroxyl groups in the Fe–SA-X. The absorption of at 1629 and 1384 cm−1 could be assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration and C–H symmetric deformation vibration. With the increase of the temperature, the intensity of C–H decreased, whereas the O–H remained unchanged. For the two samples, the bands at 561 cm−1 can be due to Fe–O. This is illustrated that Fe3O4 nanoparticles have been inserted onto the surface of carbon matrix successfully. However, with the increase of the temperature, the intensity of Fe–O bond became weaker and weaker; this indicated that part of preformed Fe3O4 was gradually reduced during the thermal treatment.
C stretching vibration and C–H symmetric deformation vibration. With the increase of the temperature, the intensity of C–H decreased, whereas the O–H remained unchanged. For the two samples, the bands at 561 cm−1 can be due to Fe–O. This is illustrated that Fe3O4 nanoparticles have been inserted onto the surface of carbon matrix successfully. However, with the increase of the temperature, the intensity of Fe–O bond became weaker and weaker; this indicated that part of preformed Fe3O4 was gradually reduced during the thermal treatment.
 |
| | Fig. 4 FTIR spectra of (a) Fe–SA-600, (b) Fe–SA-800. | |
N2 adsorption–desorption isotherms and pore size distribution of four carbonization treatment material and pre-oxidized materials described in Fig. S1.† It four samples revealed the BET surface area was 311, 342, 357, 404 m2 g−1, respectively. Detailed data were summarized in Table 1. Clearly, these Fe–SA-X (X = 600, 700, 800 and 900) has a relatively high specific surface area and average pore diameter. Effects of temperature on structural characteristics of products following discussion is likely to come from two aspects. On one hand, the expansion of gases such as carbon dioxide and water form by carbon and iron oxide in the interaction between material at higher temperatures under a N2 atmosphere, lead to an increase in surface area.38 The other hand, the temperature increases will lead to the formation of iron nanoparticles of carbon-thermal reduction reactions, will also lead to an increase in surface area.
Table 1 The BET parameters of the Fe–SA-X
| Sample |
SBET (m2 g−1) |
Vtotal (cm−3 g−1) |
Dpeak (Å) |
| Fe–SA-600 |
311.7 |
0.45 |
5.83 |
| Fe–SA-700 |
342.2 |
0.465 |
5.44 |
| Fe–SA-800 |
356.9 |
0.47 |
5.16 |
| Fe–SA-900 |
404.3 |
0.53 |
5.27 |
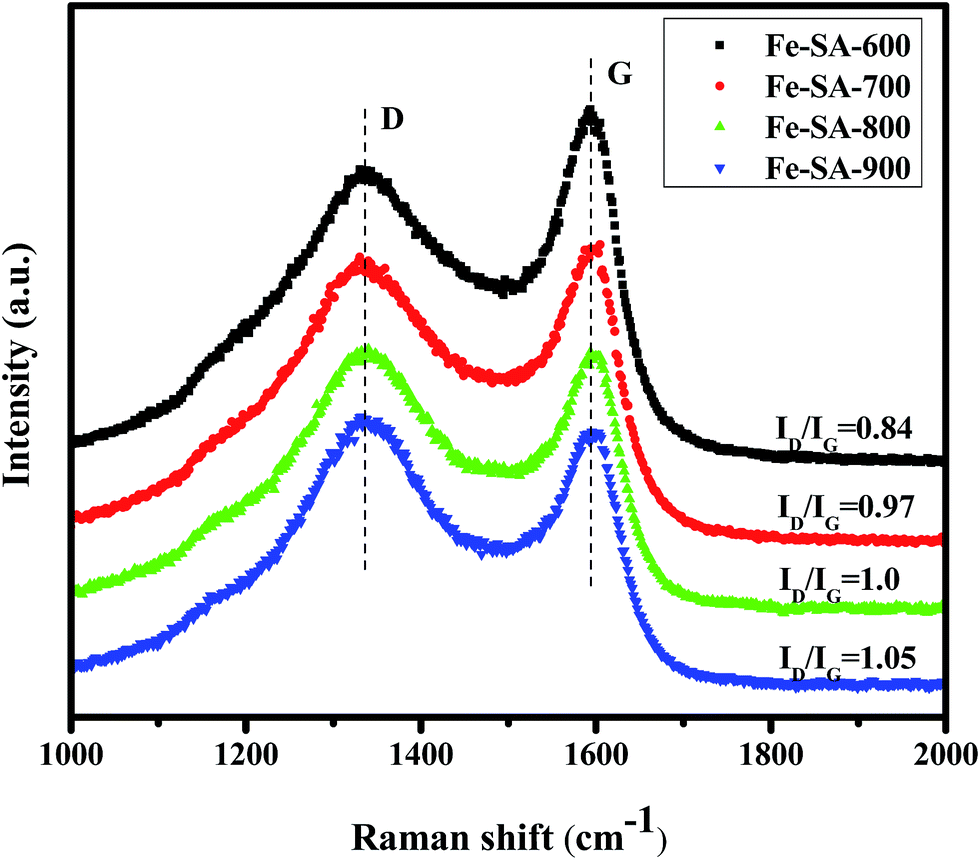
Besides, Raman spectroscopy is a powerful tool to describe the disorder or the degree of carbon. Fe–SA-X (X = 600, 700, 800 and 900) Raman spectra of samples are shown in Fig. 5. It can be clearly seen that there are two characteristic peaks, one is at 1330 cm−1, is called the D band, and the other appears at around 1593 cm−1, which is called the G band. In general, the D band arising from the defect and disorder on the graphitic lattice and the G band is assigned to the E2g C-sp2 atomic vibration modes of two-dimensional hexagonal lattice.39,40 A higher ID/IG intensity ratio is related to the graphitization degree to the amorphous state. In Fig. 5, it can be observed that the ID/IG ratios of these Fe–SA-900 composites much higher than that of the Fe–SA-600, it may be attributed to the highly disorder of the carbon matrix. For example, the largest ID/IG Fe–SA-900 ratio was 1.05, while for the corresponding Fe–SA-600 it is only 0.84. The enhanced ID/IG values was contributed to the strong D band, show that an increasing number of defects generated by high temperature carbonization. These defects on the surface of the carbon matrix will act as the dipoles, and are greatly influence the polarization. Besides, it can also be found that both D and G peaks of composites exhibit slight left shifting when compared with the Fe–SA-600. This phenomenon can be attributed to the transfer charger interface between carbon and iron.41
 |
| | Fig. 5 Raman spectra of Fe–SA-X (X = 600, 700, 800, 900). | |
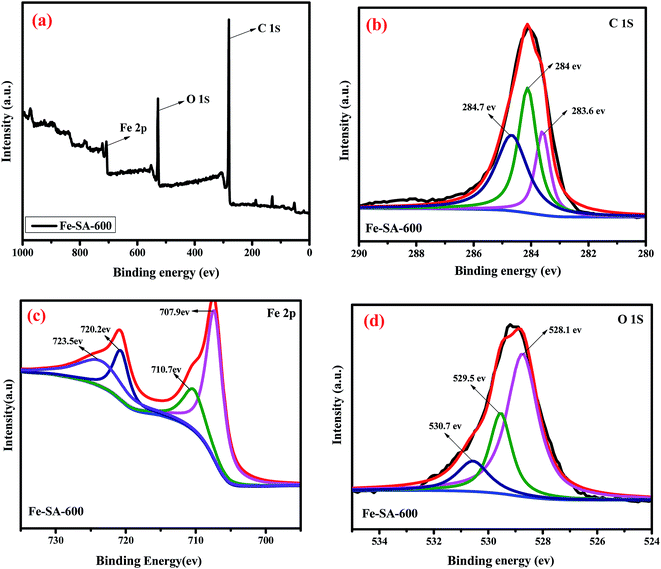
To further investigated the chemical composition and contents in the samples, XPS measurement is a powerful tool to address this issue. XPS measurements and the corresponding results were presented in Fig. 6. Three characteristic peaks: C 1s, O 1s and Fe 2p were observed clearly in the spectrum (Fig. 6a). Fig. 6a shows the survey spectra of Fe–SA-600, C (81.19 at%), O (16.04 at%) and Fe (2.76 at%) in the sample. Fig. 6b depicts the curve of C 1s in the binding energy range of 280–290 eV, three fitting peaks were observed. These peaks indicated three different species of C in the Fe–SA-600 composites. The fitting peaks at 284, 284.7 and 285.3 eV ascribed to the CC, C–H and C–O respectively.42 To our knowledge, C–H groups were the strongest reducing groups on the carbon surface.43 Because the X-ray diffraction peaks of γ-Fe2O3 are extremely similar to those of Fe3O4, XPS measurement is necessary to ensure the valence states of the Fe element. In the Fe 2p spectra (Fig. 6c), which clearly revealed Fe 2p1/2 and Fe 2p3/2 of the Fe level. The Fe 2p peak positions located at 710.7 eV and 723.5 eV corresponded to Fe 2p3/2 and Fe 2p1/2 clearly indicating the appearance of Fe3O4, this demonstrates that the composition of nanoparticles is Fe3O4. The binding energy at 707.9 eV could be related to Fe3C peak of Fe 2p3/2, and the binding energy 720.1 eV could be assigned to Fe peak of Fe 2p1/2. Besides, for the O 1s level of the Fe–SA-600 composites in Fig. 6d, the peaks situated at 528.1, 529.5 and 530.7 eV corresponded to the bonds of C–O, Fe–O and O–H respectively.
 |
| | Fig. 6 XPS spectra of Fe–SA-600, (a) survey spectra, (b) C 1s peaks, (c) Fe 2p peaks, (d) O 1s. | |
According to various characterization results, it can be found that, when the temperature rose to 900 °C from 600 °C, the main magnetic component evolved into Fe3C from preformed Fe3O4 species; and meanwhile, increased carbonization temperature made its internal structure undergone a significant change. At 600 °C, it has regular channel and uniform particle size distribution; while at higher 900 °C, although the specific surface area increased, along with the heterogeneity of pore system and structural slippage, the particle size and the distribution of metal-based component became heterogeneous. When the electromagnetic wave passed through the material, it would be detrimental to their further transmission within this kind of matrices, leading to the loss of the electromagnetic waves. Meanwhile, the reduction of the amount of Fe3O4 in this process might also decrease the electromagnetic wave loss. Accordingly, the one, i.e. Fe–SA-600, would possibly exhibit excellent electromagnetic wave absorption performance.
3.2. Magnetic properties
All above characterization results have demonstrated that the porous Fe–SA-X composites are successfully fabricated by the carbonization of the Fe–SA precursors under N2. As a type of carbon-based materials, various carbon nanostructures, such as carbon nanotubes (CNTs), carbon nanofibers (CNFs) and carbon nanospheres, have been widely used for electromagnetic wave absorption. It has been demonstrated from previous studies that the electromagnetic wave absorption performance of carbon nanostructures can be strengthen by the incorporation of iron-based materials.15–17 Considering the composite constituent and porous architecture, the composites Fe–SA-X, as-prepared, are certainly anticipated to exhibit excellent absorption capability of electromagnetic wave.
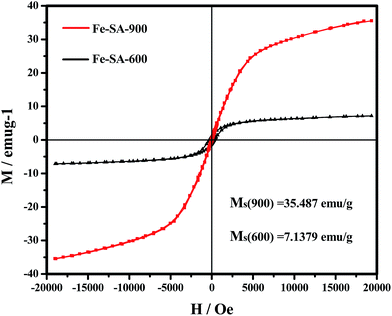
In the first step, the field-dependent magnetizations for Fe–SA-X composites were studied at room temperature using a VSM. As shown in Fig. 7, two samples showed typical ferromagnetic hysteresis loops. Obviously, the magnetization values mainly attributed to the presence of Fe species in these two samples. As we know, magnetized highly reduces the electromagnetic decay due to the obvious nature of the magnetic loss behavior. In order to better understand this effect, the following equation:44
| | |
μ′ = 1 + (M/H) cosσ
| (3) |
| | |
μ′′ = (M/H) sinσ
| (4) |
where
M is the magnetization,
H is the external magnetic field,
σ is the phase lag angle of magnetization behind an external magnetic field. In general, a high magnetization value is attributed to a strong
μ′ and
μ′′ value. In this present study, the magnetization only comes from Fe, Fe
3C and Fe
3O
4. The saturation magnetization (
MS), coercivity (
HC), and remnant magnetization
Mr are 7.1379 emu g
−1, 292.62 Oe, and 1.1321 emu g
−1 for the Fe–SA-600, and 35.487 emu g
−1, 47.745 Oe, and 0.3889 emu g
−1 for the Fe–SA-900. The low
MS of the as-prepared Fe–SA-600 samples is mainly attributed to the lower temperature carbonization, resulting in the relatively poor crystallinity of the as-prepared Fe–SA-600 samples is probably important factor that can induce the enhancement of spin disorder, thus resulting in low saturation magnetization. The saturation magnetization of Fe–SA-
X composites tends to increase with carbonization temperature. This is due to the crystallinity of the sample and magnetic properties of metal nano-particle size became larger as the carbonization temperature increases. These factors lead to the highest
MS of Fe–SA-900. Coercive (
Hc) is another key to determine the best location of the reflection loss peaks. In fact, it's hard to get optimal absorption peaks towards higher frequencies due to the lower
Hc value can be expressed by the following equation:
45| | |
tm = nc/4fm(εr − μr)1/2
| (8) |
where
μ0 stands for the universal value of permeability in free space (4π × 10
−7 H m
−1),
r represents gyromagnetic ratio, and
Ha means the anisotropy energy,
tm and
fm is the matching thickness and frequency of the RL
min peaks. For most magnetic materials,
Hc value is very low, is always less than 100 Oe called a soft magnetic material. Reduce the
Hc value will get a smaller
Ha value and lead to
fr value is low. Based on
eqn (8), a small value of
fr is not conducive to a thin coating thickness (
t).
 |
| | Fig. 7 Magnetization curves of Fe–SA-600 and Fe–SA-900, composites at room temperature (the inset is magnetization curves from −2000 to 2000 Oe). | |
3.3. Electromagnetic wave absorption properties
It can be known from the electromagnetic energy conversion principle24 that the reflection and attenuation characteristics of microwave absorption of an absorber rely heavily on its EM parameters, the complex permittivity (εr = ε′ − jε′′) and complex permeability (μr = μ′ − jμ′′) besides. The real parts (ε′ and μ′) of εr and μr stand for the storage of electric and magnetic energy, respectively. The imaginary parts (ε′′ and μ′′) stand for the loss of electric and magnetic energy. The dielectric and magnetic dissipation factors, tanδE = ε′′/ε′ and tanδM = μ′′/μ′, respectively, offer a step of the power lost in a material against the amount of power stored.46 To study the intrinsic reason for microwave absorption properties of the Fe–SA-X composite, the EM parameters in Fig. 8 shows the real (ε′ and μ′) and imaginary (ε′′ and μ′′) parts of the complex electromagnetic parameters for the Fe–SA-X composites in the range of 2–18 GHz. As illustrated in Fig. 8a, overall, the ε′ values of Fe–SA-600 and Fe–SA-700 tend to slowly decrease with increasing frequency; the ε′ value decreases from 11.19 to 4.48 for Fe–SA-600 and from 27.03 to 10.11 for Fe–SA-700, the Fe–SA-800 tend to rapidly decrease with increasing frequency from 39.72 to 15.54. But, the ε′ value of Fe–SA-900 decreased gradually with two small fluctuations in test frequency band, at a frequency of 9.1 GHz and 16.8 GHz, respectively. According to previous literature reports, Wang et al. explained this phenomenon by the increased lagging behind of the dipole-polarization response with respect to the electric-field change at higher frequencies.47 As shown in Fig. 8b, with the carbonization temperature increases, the ε′′ value also tend to decrease continuously with increasing frequency. The ε′′ value of Fe–SA-600 shows edge fluctuation (5.1–9.4), and the ε′′ value of Fe–SA-700 also shows edge variation (14.7–22.5). Moreover, the ε′′ value of Fe–SA-800 ranges from 34.07 to 57.39, the ε′′ value of Fe–SA-900 ranges from 44.08 to 70.49, which is significantly larger than those of Fe–SA-600. Likewise, tanδE of Fe–SA-900, Fe–SA-800 and Fe–SA-X-700 is greater than those of Fe–SA-600, (Fig. 8c). This indicates that Fe–SA-700, Fe–SA-800 and Fe–SA-900, as compared to Fe–SA-600, exhibits great storage and loss capabilities for electric energy. The permittivity of the Fe–SA-X (X = 700, 800, 900) can be ascribed to the increase of electric conductivity and multiple polarization among ferromagnetic nanoparticles being efficiently isolated by carbon matrix. Generally, the permittivity values of carbon composites increase with increasing loading of metals, which exhibit higher electric conductivity.31 Among the four samples, Fe–SA-900 exhibits the high electric conductivity due to the good crystallinity of Fe3C nanoparticles. In particular, the porous structure could have decreased the effective permittivity and great impedance match.48 Fe–SA-600 have an apparent porous structure. With the increasing of carbonization temperature, composites structure is destroyed. When the carbonization temperature increased to 900 °C, the structure destroyed, and metal particles became larger, as is seen from the TEM and SEM (Fig. 1 and 2). As a consequence, Fe–SA-600 has poor effective permittivity and better impedance match.
 |
| | Fig. 8 Frequency dependence of electromagnetic parameters of the samples obtained at different temperatures. (a) The real part (ε′) and (b) imaginary part (ε′′) of complex permittivity, (c) dielectric loss (tanδE), (d) real part (μ′) and (e) imaginary part (μ′′) of complex permeability, (f) magnetic loss (tanδM). | |
The real part (μ′) and imaginary part (μ′′) of relative complex permeability of the composites are shown in Fig. 8d and e. It can clearly be seen that the μ′ values exhibit an increase tendency in the frequency of 2–18 GHz with a small fluctuation. The μ′′ values exhibit a decrease tendency in the frequency of 2–18 GHz, only the μ′′ values of the Fe–SA-900 composite increase significantly in the range of 16–18 GHz. Among the four samples, the μ′′ values are fluctuates around zero, implying the natural resonance occurred in the Fe–SA-X composite.17 The electromagnetic transformation is related to the energy transfer between vectors of electric and magnetic fields, according to previous literature, and further gives rise to the resonance behavior when their energy states satisfy the matching with the frequency of the electromagnetic wave.17 The negative values of the imaginary part show that the loss with regard to the magnetic vector is even smaller than those needed for the permeability in medium, which is probably aroused by the radiation and/or transfer of energy.49 The natural resonance equation is as followed: 2πfr = γHa, Ha = 4|K1|/3μ0MS,50 where γ represents gyromagnetic ratio, Ha means the anisotropy field, f stands for the resonance frequency and |K1| is the anisotropy coefficient. On one hand, the anisotropy energy of small materials would remarkably increase due to the surface anisotropic field by the small size effect. On the other hand, the MS value of the Fe–SA-600 composites is much lower than that of the Fe–SA-900, thus the anisotropy energy of the Fe–SA-600 composites is higher than that of the Fe–SA-900. The higher anisotropy energy is helpful for the improvement of microwave absorption properties.
As compared to the other three samples, the decrease of the relative permeability of Fe–SA-600 composite is attributed to the poor magnetization value, which due to the poor crystallinity resulted from the lower temperature carbonization. The matching between the relative permeability (μr) and permittivity (εr) is mainly reflected in the comparison of the magnetic dissipation factors (tanδM) and dielectric dissipation factors (tanδE). The difference between tanδE and tanδM of Fe–SA-900, Fe–SA-800 and Fe–SA-700 is large; contrarily, the Fe–SA-600 has closer value of tanδE and tanδM. Thus, Fe–SA-600 exhibits better impedance matching and possesses higher loss capabilities for magnetic energy. Therefore Fe–SA-600 might exhibit the excellent electromagnetic wave absorption performance.
As far as we know, the impedance matching ratio and attenuation constant α can directly determine the electromagnetic absorption performance. It is easily comprehended that by the high attenuation constant value, more and more electromagnetic wave should be allowed to travel into the absorber and then attenuate it by the absorber. According to the literature, the better of the impedance matching, the stronger of the ability of reduction, favorably leading to the absorption of electromagnetic waves over the employed substances.52 However, Fe–SA-600 has a high impedance matching ratio. The attenuation constant α means the integral attenuation effect including magnetic and dielectric loss, as calculated according to the below formula:53
| | |
α = (2πf/c)1/2 × [(μ′′ε′′ − μ′ε′) + [(μ′′ε′′ − μ′ε′)2 + (μ′′ε′′ + μ′ε′)2]1/2]1/2
| (9) |
According to the analysis, Fe–SA-600 still shows the best attenuation ability in the 13.2–18 GHz region. Its high improvement impedance matching ratio and the larger attenuation constant make it possess excellent microwave absorption properties in a small coating thickness.
In order to calculated the electromagnetic wave absorption performance of Fe–SA-X composites, the reflection loss (RL) values are calculated from the relative complex permeability (μr) and permittivity (εr) at a given frequency (2–18 GHz) and absorber thickness. Fig. 9a–d shows the calculated theoretical reflection loss (RL) of the composites-wax with different thicknesses in the frequency range of 2–18 GHz with a loading of 25 wt%. Fe–SA-600 exhibits great microwave absorption ability, and the maximum RL is −24 dB at 17 GHz with a thickness of 1.5 mm. The frequency range is quite broader about have 4.8 GHz. Noticeably, the bandwidths RL values below −10 dB (90% microwave absorption) exceeds 4.8 GHz when the composites weight ratio is 25% with thickness of 1.5 mm. These values are comparable to those reported results (Table 2), demonstrating the high-efficiency of this newly fabricated porous absorbent. With the increase of carbonization temperature, reduce the absorption properties of the samples. The decrease in amplitude follows the order of Fe–SA-600 > Fe–SA-700 > Fe–SA-800 > Fe–SA-900. Among the four samples, Fe–SA-800 and Fe–SA-900 exhibit the worst electromagnetic wave absorption property with the maximum RL less than −7 dB. However, the similar values of dielectric loss and magnetic loss can lead to the good impedance match of electromagnetic wave absorbing materials.31 Fe–SA-X (X = 900, 800, 700) has the higher dielectric loss, for which is significantly greater than its magnetic loss value. Thus, it has a poor impedance match. The magnetic loss (μ) value and dielectric loss (ε) value of Fe–SA-600 is close, indicating that Fe–SA-600 have a excellent impedance match.
 |
| | Fig. 9 Calculated results of the reflection loss vs. frequency for different samples with different thicknesses: Fe–SA-600, Fe–SA-700, Fe–SA-800 and Fe–SA-900 samples. | |
Table 2 Electromagnetic wave absorption properties of reported and studied composites
| Sample |
RL (dB) |
Thickness (mm) |
Frequency range (RL < −10 dB) (GHz) |
Ref. |
| Fe–Co/nanoporous carbon |
−21.7 |
1.2 |
12.2–18 |
51 |
| Ni/SnO2 |
−36.7 |
1.7 |
10.6–14 |
53 |
| Co/C |
−35.3 |
2.5 |
8.4–14.2 |
31 |
| RGO–Fe3O4 |
−15.3 |
2.0 |
10.4–13.2 |
54 |
| Fe–SA-600 |
−24 |
1.5 |
13.2–18 |
This work |
As mentioned above, the decent electromagnetic wave absorption performance of a material need excellent impedance match and absorption property, which are connected with complicated permittivity, complicated permeability, thickness and material structure. In Fig. 9a, the strong reflection loss peaks of Fe–SA-600 are ascribed to resonant absorption at a given frequency (2–18 GHz), which should be caused by the “structure effect”.31 Obviously, porous materials have great impedance match with a free space than solid materials due to their poor effective permittivity.50 From SEM observation (Fig. 1), compared to Fe–SA-600, Fe–SA-800 and Fe–SA-900 with the increase of temperature, structure is destroyed and carbon spheres increased. With a wealth of porous structure, these multi-interfaces result in significant interfacial polarization and multi-polarization, which would enhance the dielectric loss obviously.30 Above all, Fe–SA-600, compared to Fe–SA-700, Fe–SA-800 and Fe–SA-900, has better impedance match because of their low effective permittivity. From the above analysis, the results show that the Fe–SA-600 composites material could be used as lightweight and high efficiency EMW absorbing material.
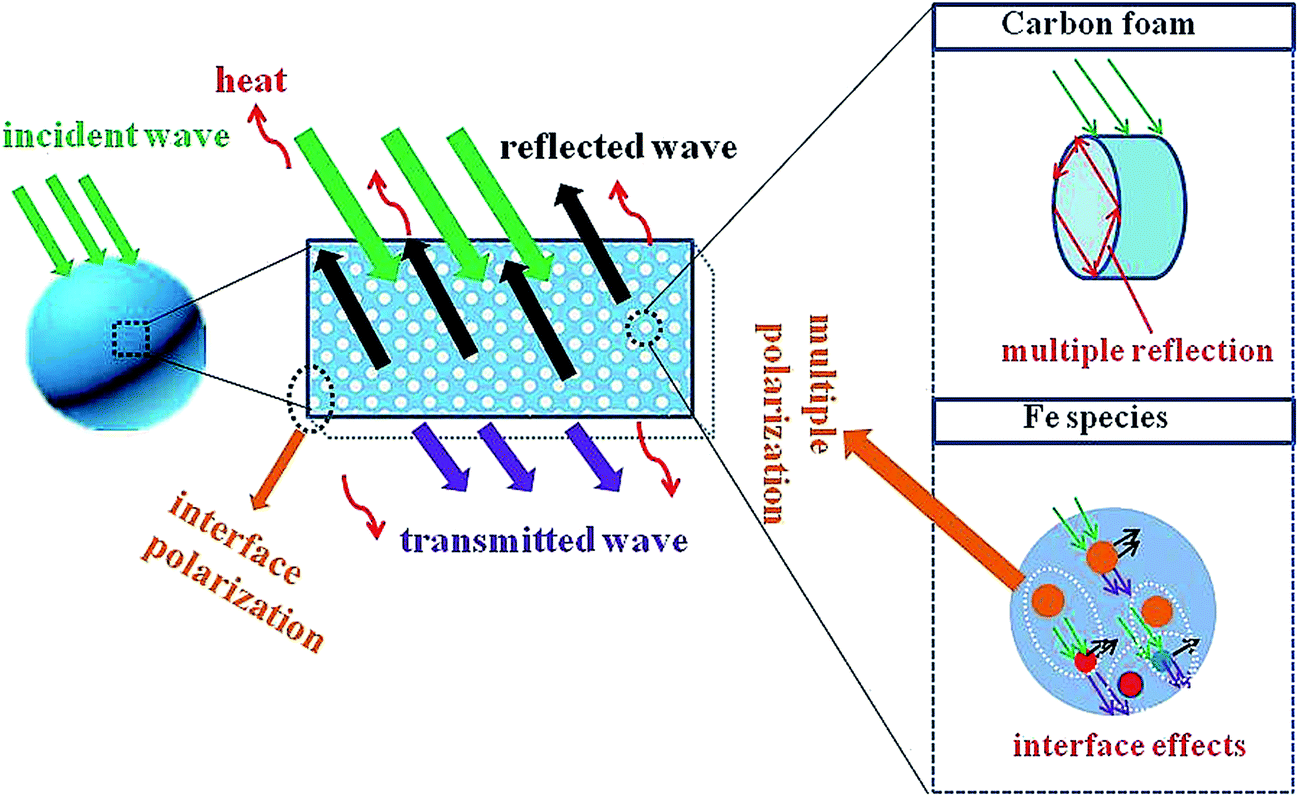
Based on the above analysis, the attenuation and impedance matching are two crucial factors, resulting in electromagnetic wave absorption properties. Therefore, we can conclude that mechanism may come from the following areas (Scheme 2). When the electromagnetic wave propagates into the absorber part of the electromagnetic energy consumption more than strong polarization between Fe3O4–Fe, Fe3O4–Fe3C and Fe3O4–C interface, and then energy is dissipated in the form of heat. Possible magnetic domains exchange (Fe3O4, Fe and Fe3C magnetic field) and natural resonance play a key role in the attenuation of electromagnetic waves. Assisted by iron, Fe3C and Fe3O4, part of the electromagnetic wave is converted to carbon surface on the clinical matrix.55 Defective surface carbon matrix as a dipole center may increase the resistivity. Therefore, the clinical will be attenuated form. The nuances of electromagnetic absorption properties of Fe–SA-60–Fe–SA-900 is mainly of carbonization temperature-related. Different carbonization temperature may lead to different surface area and surface energy, resulting in different surface conditions and ID/IG values. It will then lead to different carbon matrix, which shows different attenuation.
 |
| | Scheme 2 Possible mechanism for the microwave absorption on the surface of Fe–SA-600 samples. | |
4. Conclusions
In summary, we have developed a novel Fe–SA-X composite with an enhanced microwave absorption performance using a combination of controllable cross-linking and carbonization methods. Our experiments demonstrated that the resultant porous Fe–SA-X composites exhibited great electromagnetic wave absorption properties, due to the synergetic effects between the multiple component and highly porous structure. The maximum RL of Fe–SA-600, among the four Fe–SA-X (X = 600, 700, 800, 900 °C) composites synthesized at different temperatures, reached −24 dB at 17 GHz with a thickness of 1.5 mm, and the effective absorption bandwidth (RL ≤ −10 dB) was 4.80 GHz (13.2–18 GHz). This synthetic method for Fe–SA-X absorbers is quite facile, and abundant biomass materials can provide possibilities for fabricating other porous metal/carbon composites with a wide range of compositions. The integration of light-weight, low-cost, bio-gradation and good chemical stability of composites make it promising candidates for microwave absorption.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21546008, 21676039), the Program for Liaoning Innovative Research Team in University (LT2013012) and the Program for Liaoning Excellent Talents in University (LJQ2014056) is highly appreciated.
References
- H. J. Wu, G. L. Wu, Y. Y. Ren, L. Yang, L. D. Wang and X. H. Li, J. Mater. Chem. C, 2015, 3, 7677–7690 RSC.
- Z. H. Yang, Z. W. Li, L. H. Yu, Y. H. Yang and Z. C. Xu, J. Mater. Chem. C, 2014, 2, 7583–7588 RSC.
- L. Wang, Y. Huang, X. Sun, H. J. Huang, P. B. Liu, M. Zong and Y. Wang, Nanoscale, 2014, 6, 3157–3164 RSC.
- X. Ding, Y. Huang and J. G. Wang, RSC Adv., 2015, 5, 64878–64885 RSC.
- H. T. Guan, J. B. Xie, G. Chen and Y. D. Wang, Mater. Chem. Phys., 2014, 143, 1061–1068 CrossRef CAS.
- R. L. Ji, C. B. Cao, Z. Chen, H. Z. Zhai and J. Bai, J. Mater. Chem. C, 2014, 2, 5944–5953 RSC.
- Z. X. Yu, Z. P. Yao, N. Zhang, Z. J. Wang, C. X. Li, X. J. Han, X. H. Wu and Z. H. Jiang, J. Mater. Chem. A, 2013, 1, 4571–4576 CAS.
- J. Joo and C. Y. J. Lee, J. Appl. Phys., 2000, 88, 513–518 CrossRef CAS.
- A. Wang, W. Wang, C. Long, W. Li, J. Guan, H. Gu and G. Xu, J. Mater. Chem. C, 2014, 2, 3769–3776 RSC.
- X. Huang, J. Zhang, M. Lai and T. Sang, J. Alloys Compd., 2015, 627, 367–373 CrossRef CAS.
- Y. Yang, C. Xu, Y. Xia, T. Wang and F. Li, J. Alloys Compd., 2010, 493, 549–552 CrossRef CAS.
- B. Bateer, L. Wang, L. Zhao, P. Yu, C. G. Tian, K. Pan and H. G. Fu, RSC Adv., 2015, 5, 60135–60140 RSC.
- M. Zong, Y. Huang, Y. Zhao, X. Sun, C. Qu, D. Luo and J. Zheng, RSC Adv., 2013, 3, 23638–23648 RSC.
- M. K. Han, X. W. Yin, L. Kong, M. Li, W. Y. Duan, L. T. Zhang and L. F. Cheng, J. Mater. Chem. A, 2014, 2, 16403–16409 CAS.
- R. B. Yang, P. M. Reddy, C. J. Chang, P. A. Chen, J. K. Chen and C. C. Chang, Chem. Eng. J., 2016, 285, 497–507 CrossRef CAS.
- Y. F. Pan, G. S. Wang and Y. H. Yue, RSC Adv., 2015, 5, 71718–71723 RSC.
- B. H. Bateer, L. Wang, L. Zhao, P. Yu, C. G. Tian, K. Pan and H. G. Fu, RSC Adv., 2015, 5, 60135–60140 RSC.
- L. F. Chen, Z. H. Huang, H. W. Liang, H. L. Gao and S. H. Yu, Adv. Funct. Mater., 2014, 24, 5104–5111 CrossRef CAS.
- W. E. Tenhaeff, O. Rios, K. More and M. A. McGuire, Adv. Funct. Mater., 2014, 24, 86–94 CrossRef CAS.
- S. X. Wang, L. Yang, L. P. Stubbs, X. Li and C. He, ACS Appl. Mater. Interfaces, 2013, 5, 12275–12282 CAS.
- W. Zhao, P. Yuan, X. L. She, Y. Z. Xia, S. Komarneni, K. Xi, Y. K. Che, X. D. Yao and D. J. Yang, J. Mater. Chem. A, 2015, 3, 14188–14194 CAS.
- D. H. Li, C. X. Lv, L. Liu, Y. Z. Xia, X. L. She, S. J. Guo and D. J. Yang, ACS Cent. Sci., 2015, 1, 261–269 CrossRef CAS PubMed.
- E. Fourest and B. Volesky, Appl. Biochem. Biotechnol., 1997, 67, 215–226 CrossRef CAS.
- I. Braccini and S. Perez, Biomacromolecules, 2001, 2, 1089–1096 CrossRef CAS PubMed.
- L. Liu, X. F. Yang, C. X. Lv, A. M. Zhu, X. Y. Zhu, S. J. Guo, C. M. Chen and D. J. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 7047–7053 CAS.
- Y. H. Zou, X. F. Yang, C. X. Lv, T. C. Liu, Y. Z. Xia, L. Shang, D. J. Yang and T. R. Zhang, Adv. Sci., 2016, 9, 1–8 Search PubMed.
- L. Liu, X. F. Yang, N. Ma, H. T. Liu, Y. Z. Xia, C. M. Chen, D. J. Yang and X. D. Yao, Small, 2016, 5, 1295–1301 CrossRef PubMed.
- Y. H. Zou, S. Chen, X. F. Yang, N. Ma, Y. Z. Xia, D. J. Yang and S. J. Guo, Adv. Energy Mater., 2016, 9, 1–7 Search PubMed.
- M. Toru, S. Satoshi, K. Toshio, T. Nobuki and I. Koichiro, J. Magn. Magn. Mater., 2004, 281, 195–205 CrossRef.
- X. L. Li, Y. X. Qi, Y. F. Li, Y. Zhang, X. H. He and Y. H. Wang, Bioresour. Technol., 2013, 142, 611–619 CrossRef CAS PubMed.
- Y. Y. Lü, Y. T. Wang, H. L. Li, Y. Lin, Z. Y. Jiang, Z. X. Xie, Q. Kuang and L. S. Zheng, ACS Appl. Mater. Interfaces, 2015, 7, 13604–13611 Search PubMed.
- Z. M. Lei, Q. D. An, Y. Fan, J. L. Lv, C. Gao, S. R. Zhai and Z. Y. Xiao, New J. Chem., 2016, 40, 1195–1204 RSC.
- C. Y. Cao, J. Qu, W. S. Yan, J. F. Zhu, Z. Y. Wu and W. G. Song, Langmuir, 2012, 28, 4573–4579 CrossRef CAS PubMed.
- L. B. Cui, Y. Liu, X. H. Wu, Z. Q. Hu, Z. J. Shi and H. J. Li, RSC Adv., 2015, 5, 75817–75822 RSC.
- H. Guo, S. F. Zhang, Z. N. Kou, S. R. Zhai, W. Ma and Y. Yang, Carbohydr. Polym., 2015, 115, 177–185 CrossRef CAS PubMed.
- J. Zhu, H. Gu, J. Guo, M. Chen, H. Wei, Z. Luo, H. A. Colorado, N. Yerra, D. Ding, T. C. Ho, J. Hopper, D. P. Young, Z. Guo and S. Wei, J. Mater. Chem. A, 2014, 2, 2256–2265 CAS.
- B. Qiu, J. Guo, X. Zhang, D. Z. Sun, H. B. Gu, Q. Wang, H. W. Wang, X. F. Wang, X. Zhang, B. L. Weeks, Z. H. Guo and S. Y. Wei, ACS Appl. Mater. Interfaces, 2014, 6, 19816–19824 CAS.
- Z. M. Lei, S. R. Zhai, J. L. Lv, Y. Fan, Q. D. An and Z. Y. Xiao, RSC Adv., 2015, 5, 77932–77941 RSC.
- T. Palaniselvam, M. O. Valappil, R. Illathvalappil and S. Kurungot, Energy Environ. Sci., 2014, 7, 1059–1067 CAS.
- B. Wen, M. S. Cao, M. M. Lu, W. Q. Cao, H. L. Shi, J. Liu, X. X. Wang, H. B. Jin, X. Y. Fang, W. Z. Wang and J. Yuan, Adv. Mater., 2014, 26, 3484–3489 CrossRef CAS PubMed.
- M. Mishra, A. P. Singh, B. P. Singh, V. N. Singh and S. K. Dhawan, J. Mater. Chem. A, 2014, 2, 13159–13168 CAS.
- H. Chen, X. X. Wang, J. X. Li and X. K. Wang, J. Mater. Chem. A, 2015, 3(11), 6073–6081 CAS.
- S. X. Chen and H. M. Zeng, Carbon, 2003, 41, 1265–1271 CrossRef CAS.
- B. Lu, H. Huang, X. L. Dong, X. F. Zhang, J. P. Lei, J. P. Sun and C. Dong, J. Appl. Phys., 2008, 104, 114313 CrossRef.
- H. L. Lv, X. H. Liang, Y. Cheng, H. Q. Zhang, D. M. Tang, B. S. Zhang, G. B. Ji and Y. W. Du, ACS Appl. Mater. Interfaces, 2015, 7, 4744–4750 CAS.
- X. Li, B. Zhang, C. Ju, X. Han, Y. Du and P. Xu, J. Phys. Chem. C, 2011, 115, 12350–12357 CAS.
- H. Wang, H. H. Guo, Y. Y. Dai, D. Y. Geng, Z. Han, D. Li, T. Yang, S. Ma, W. Liu and Z. D. Zhang, Appl. Phys. Lett., 2012, 101, 083116 CrossRef.
- L. L. Diandra and D. R. Reuben, Chem. Mater., 1996, 8, 1770 CrossRef.
- J. L. Lv, S. R. Zhai, C. Gao, N. Zhou, Q. D. An and B. Zhai, Chem. Eng. J., 2016, 289, 261–269 CrossRef CAS.
- B. Wang, J. Zhang, T. Wang, L. Qiao and F. Li, J. Alloys Compd., 2013, 567, 21–25 CrossRef CAS.
- X. M. Zhang, G. B. Ji, W. Liu, B. Quan, X. H. Liang, C. M. Shang, Y. Cheng and Y. W. Du, Nanoscale, 2015, 7, 12932–12942 RSC.
- H. L. Lv, G. B. Ji, W. Liu, H. Q. Zhang and Y. W. Du, J. Mater. Chem. C, 2015, 3, 10232–10242 RSC.
- B. Zhao, G. Shao, B. B. Fan, W. Y. Zhao and R. Zhang, RSC Adv., 2014, 4, 57424–57429 RSC.
- X. Sun, J. P. He, G. X. Li, J. Tang, T. Wang, Y. X. Guo and H. R. Xue, J. Mater. Chem. C, 2013, 1, 765–777 RSC.
- H. L. Lv, G. B. Ji, X. H. Liang, H. Q. Zhang and Y. W. Du, J. Mater. Chem. C, 2015, 3, 5056–5064 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22721j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.