DOI:
10.1039/C6RA22693K
(Paper)
RSC Adv., 2016,
6, 108708-108716
Synthesis, characterization and biological activity of fluorescently labeled bedaquiline analogues†
Received
11th September 2016
, Accepted 1st November 2016
First published on 2nd November 2016
Abstract
Diarylquinolines represent a new class of antibiotics with high potency against Mycobacterium tuberculosis. As such, they are of utmost importance in the treatment of drug-resistant bacterial pathogens. In this work, we report a strategy for preparing fluorescently labeled derivatives of the FDA-approved diarylquinoline-based tuberculosis drug bedaquiline. The labeled compounds were capable of blocking bacterial growth and interfered with the function of ATP synthase, the cellular target of diarylquinolines. This indicates that the chosen labeling strategy does not preclude the antibacterial activity of bedaquiline, and allowed us to investigate the effect of labeling on drug recognition by bacterial efflux pumps in living M. tuberculosis strains. These properties, coupled with the efficient fluorescence of the attached BODIPY fluorophore means that these compounds can be used as a research tool to gain deeper understanding into the mechanism of action of this class of drugs.
Introduction
In 2014, some 9 million individuals were newly infected worldwide with Mycobacterium tuberculosis, and ca. 1.5 million patients died from tuberculosis.1 In particular, multi-drug resistant and extremely drug resistant strains of M. tuberculosis are unyielding to treatment with current tuberculosis chemotherapy.2 Consequently, new drugs are urgently needed to counteract the development of drug resistance. The use of small molecules that inhibit energy metabolism is a new approach to kill M. tuberculosis and related pathogenic mycobacteria.3 Several new or repurposed drug entities are known to block the oxidative phosphorylation pathway, a key route in energy metabolism. Clofazimine and related phenothiazines interfere with the type-II NADH dehydrogenase function,4 the imidazopyridines inhibit the cytochrome bc1 complex,5 and the diarylquinolines inhibit ATP synthase in mycobacteria and other Gram-positive pathogenic bacteria.6 Bedaquiline, the lead compound of the diarylquinolines, is a stereo-specific ATP synthase inhibitor7 which acts strongly bactericidal against M. tuberculosis.8 Bedaquiline has been approved by the US Food and Drug Administration and by the European Medicines Agency for treatment of multi-drug resistant tuberculosis. Bedaquiline and other drugs targeting energy metabolism may well contribute to shortening and simplifying tuberculosis chemotherapy.9
However, a detailed understanding of the mechanism of action of bedaquiline is required in order to fully exploit its potential in chemotherapy, and perhaps develop novel bedaquiline analogues. Utilization of a fluorescent drug derivative can be a valuable strategy to study antibacterial drugs, as reported earlier for the characterization of the benzothiazidinone class of drugs.10 Two prerequisites must be fulfilled for successful implementation of drug labeling in such investigations, i.e., that the fluorescence of the label is not quenched by interactions with the drug or the environment, and that the antibacterial action and target binding of the drug is not precluded by the presence of the label.
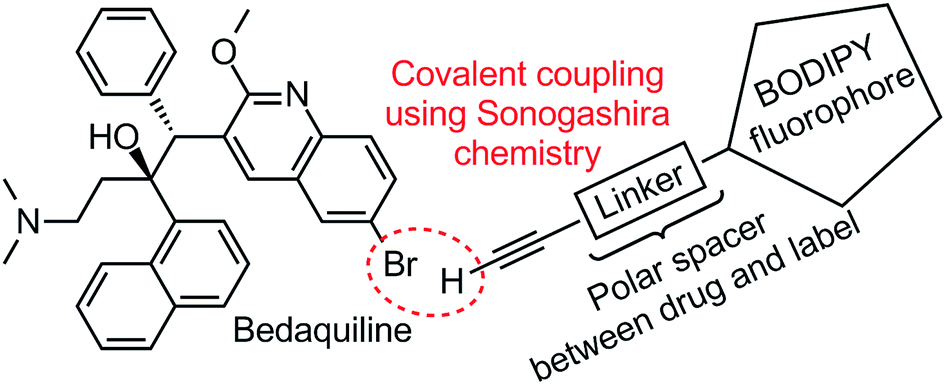
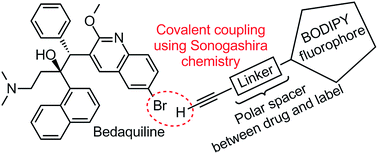
In this study we report on the design and execution of a labeling strategy for bedaquiline, by covalently attaching a BODIPY fluorophore to the quinoline ring system of bedaquiline (see Fig. 1). The synthesis and photochemical characterization of two such fluorescent derivatives is described, and their antibacterial activity is determined. This specific fluorescent label is chosen by virtue of the compact size, photochemical stability and ease of chemical synthesis and modification of BODIPY-type compounds.11 A linear polar spacer of variable length is used to separate the drug and the label, with the aim of keeping bactericidal activity intact and the fluorescence of the label unperturbed by the coupling to bedaquiline. Considering the importance of creating labeled compounds that are sufficiently similar to the parent compound under investigation, we also evaluated if the newly prepared compounds share affinity for the same drug efflux pump system in M. tuberculosis as the parent compound.
 |
| | Fig. 1 Structural formula of bedaquiline and schematic depiction of fluorescent label. Using a Sonogashira cross-coupling, the alkyne-terminated polar linker attached to the BODIPY fluorophore will be covalently coupled to the aryl bromide position of bedaquiline. | |
Materials and methods
Assay of ATP synthesis by mycobacterial membrane vesicles
Inverted membrane vesicles were prepared from M. smegmatis12 and ATP synthesis was measured as described earlier.13 Briefly, membrane vesicles (1 mg mL−1) from M. smegmatis were incubated in 50 mM morpholino-ethanesulfonic acid (MES, pH 6.5, 5 mM MgCl2), 2 mM ADP, 20 mM KH2PO4, 100 μM P1,P5-di(adenosine-5′) pentaphosphate (Ap5A), 25 mM glucose, 11.8 U mL−1 hexokinase (Sigma), and protease inhibitors (complete, EDTA-free; protease inhibitor cocktail tablets from Roche). Samples (0.25 mL) were incubated at 37 °C with vigorous stirring in 18 mL flasks. The reaction was initiated with 5 mM succinate. After 2 hours, each reaction was stopped with 25 mM EDTA, followed by transfer to ice. Samples were transferred to Eppendorf tubes, boiled for 5 min, and centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 20 min) to remove denatured protein. In supernatants, the synthesized glucose-6-phosphate was quantified by NADP+ (2.5 mM) reduction in the presence of 3 U mL−1 of glucose-6-phosphate dehydrogenase (Roche). NADPH formation was monitored with a spectrophotometer at 340 nm.
000g, 20 min) to remove denatured protein. In supernatants, the synthesized glucose-6-phosphate was quantified by NADP+ (2.5 mM) reduction in the presence of 3 U mL−1 of glucose-6-phosphate dehydrogenase (Roche). NADPH formation was monitored with a spectrophotometer at 340 nm.
MIC determination by resazurin microtiter assay
The MIC of bodipy-labeled bedaquiline compounds against M. tuberculosis in the presence or absence of the efflux inhibitor verapamil (at 40 μg mL−1) was measured by the resazurin microtiter assay (REMA).14 Serial 2-fold dilutions of antibiotics were performed in 7H9/0.5% glycerol/10% OADC (7H9 broth), in 96-well microtiter plates, with a final volume of 100 μL per well. A volume of 100 μL of inoculum suspension was added to each well. For preparation of this inoculum either liquid cultures in logarithmic phase were adjusted to OD600 ∼ 0.3 and diluted 1/100 in 7H9 broth. Plates were incubated 7 days at 37 °C. To each well, 40 μL of a resazurin solution 0.1 mg mL−1 were added, and plates were incubated for an additional 48 hours at 37 °C. Resazurin is an indicator of bacterial growth; as a consequence of the metabolic activity of bacteria, resazurin (blue color, weakly fluorescent) is reduced to resorufin (pink color, fluorescent). Interpretation of results was based on visual inspection of change of color (blue to pink). MIC was defined as the lowest concentration of antibiotic that prevented growth.
Synthetic protocols; general information
CH2Cl2 (HPLC grade) was obtained from VWR and used as received. Anhydrous N,N-dimethylformamide (DMF) was obtained from Sigma-Aldrich and used as received. THF was distilled from sodium/potassium alloy and stored in a Schlenk flask before use. Solvents used for chromatography were of HPLC or spectrographic grade. All other reagents were obtained from Sigma-Aldrich or VWR and used as received. Column chromatography was performed on Merck “Type E” aluminum oxide (particle size 150 mesh) or SiliCycle silica gel (SiO2, 40–63 μm particles) using the described eluent systems. Thin Layer Chromatography (TLC) was performed using TLC plates from Merck (Al2O3: “Neutral F60” with F254 indicator on aluminium backing. SiO2: “Kieselgel F60” with F254 indicator on aluminium backing) and compounds were visualized by visual inspection and UV detection at 254/366 nm. Melting points were measured using ΔT = 1 °C min−1 on a Stuart Scientific SMP3 melting point apparatus on the temperature range 50–400 °C, and are uncorrected. Boiling points were measured in a similar manner, using an open-ended boiling point tube. Infrared (IR) spectra were recorded neat using a Shimadzu FTIR-8400s spectrophotometer and wavelengths are reported in cm−1. 1H, HSQC, HMBC and 13C (Attached Proton Test) nuclear magnetic resonance (NMR) spectra were used to characterize prepared compounds, and were recorded on a Bruker Avance 500 (125.78 MHz for 13C) or Bruker Avance 400 (100.64 MHz for 13C) using the residual solvent as internal standard. 19F spectra were recorded on a Bruker 250 spectrometer without any calibration. Electrospray ionisation (ESI) mass spectrometry was carried out using a Bruker micrOTOF-Q instrument in positive ion mode (capillary potential of 4500 V). Electron Impact (EI) mass spectrometry was carried out using a Shimadzu GCMS-QP2010 Plus. Determination of absorption and emission spectra and the related parameters was performed using a Shimadzu UV-2501PC spectrophotometer (absorption measurements) and a Jobin Yvon Fluorolog spectrophotometer (fluorescence measurements). Bedaquiline fumarate was obtained from Johnson & Johnson; the free base used during synthesis was prepared by extracting a CH2Cl2 solution of the fumarate three times with saturated NaHCO3 solution. The identity and purity of the free base thus afforded from the material supplied by Johnson & Johnson was verified using HRMS, chiral HPLC and multinuclear NMR spectroscopy.
Synthesis of 4-(but-3-yn-1-yloxy)benzaldehyde (A1)
To dry THF (200 mL) kept at 0 °C under an N2 atmosphere was added 3-butyn-1-ol (1.00 g, 8.20 mmol, 1.0 eq.), triphenylphosphine (2.26 g, 8.60 mmol, 1.05 eq.) and 4-hydroxybenzaldehyde (574 mg, 8.20 mmol, 1.0 eq.).19 After five minutes of stirring, diethylazodicarboxylate (DEAD; 2.2 M toluene solution, 3.73 mL, 8.20 mmol) was added via syringe. The solution was warmed up to room temperature overnight; a color change from yellow to colorless was observed. After 48 h, the solution was concentrated in vacuo. Column chromatography (SiO2 stationary phase, gradient elution 5% EtOAc in cyclohexane to 15% EtOAc in cyclohexane) afforded 4-(but-3-yn-1-yloxy)benzaldehyde (354 mg, 2.03 mmol, 25%) as a white crystalline solid. TLC (4:1 cyclohexane:EtOAc): Rf = 0.50, visualized using 254 nm light. 1H NMR (500 MHz, DMSO): δ 9.87 (s, 1H, –CHO), 7.86 (d, 2H, 3J(H,H) = 8.5 Hz, CH), 7.14 (d, 2H, 3J(H,H) = 8.5 Hz, CH), 4.18 (t, 2H, 3J(H,H) = 6.4 Hz, –OCH2–), 2.92 (t, 1H, 4J(H,H) = 2.5 Hz, –CCH), 2.68 (dt, 2H, 4J(H,H) = 2.5 Hz, 3J(H,H) = 6.4 Hz, –CH2CC–) ppm. 13C NMR (125.8 MHz, DMSO, 293 K): δ 191.4 (–CHO), 163.2, 131.9, 129.8, 115.0, 81.19 (–CCH), 72.7 (–CCH), 66.2 (–OCH2–), 18.8 (–CH2CC–) ppm. HRMS (ESI): calc. [M + H]+: 175.0754; found 175.0746. MS (EI+ mode): calc. [M]+: 174.1; found 174.1. Calc. [M–CHO]+: 145.1; found 145.1. Calc. [M-O(CH2)2CCH]+: 121.0; found 121.0. IR: 3209, 2824, 2806, 2745, 1672, 1601, 1576, 1529, 1508, 1393, 1250, 1215, 1163, 1109, 1038, 893, 827, 729, 652 cm−1. MP: 68–70 °C.
Synthesis of but-3-yn-1-yloxy-BODIPY (B1)
To dry THF (100 mL) under an N2 atmosphere was added aldehyde A1 (300 mg, 1.73 mmol, 1.0 eq.), 2,4-dimethylpyrrole (362 mg, 3.81 mmol, 2.2 eq.) and 2 drops of trifluoroacetic acid. After 16 hours of stirring at room temperature, DDQ (398 mg, 1.73 mmol, 1.0 eq.) was added as a solution in 10 mL THF. After 1 hour of stirring, the reaction mixture was cooled to 0 °C using ice, and NEt3 (2.0 mL, 14.3 mmol, ∼8 eq.) was added via syringe followed by BF3·OEt2 (2.5 mL, 25.3 mmol, ∼15 eq.). The reaction mixture was allowed to warm up to room temperature over the course of two hours, after which it is filtered over celite. The celite is washed with 100 mL CH2Cl2; pooled organic phases are concentrated in vacuo. The brown, sticky solid thus obtained is diluted with 100 mL of EtOAc and washed with satd. NaHCO3, satd. NH4Cl, water and brine. After drying over Na2SO4,the organic layer is concentrated in vacuo. Column chromatography (SiO2 stationary phase, gradient elution cyclohexane to 20% EtOAc in cyclohexane) afforded but-3-yn-1-yloxy-BODIPY B1 (335 mg, 0.38 mmol, 22%) as a orange crystalline solid. TLC (2:1 cyclohexane:EtOAc): Rf = 0.85, red spot with green fluorescence. 1H NMR (500.2 MHz, CDCl3, 293 K): δ 7.17 (d, 2H, 3J(H,H) = 8.5 Hz, phenyl CH), 7.12 (d, 3J(H,H) = 8.5 Hz, 2H, phenyl CH), 5.98 (s, 2H, pyrrole CH), 4.16 (t, 3J(H,H) = 7.0 Hz, 2H, –OCH2–), 2.74 (dt, 2H, 4J(H,H) = 2.0 Hz, 3J(H,H) = 7.0 Hz, –CH2CC–), 2.55 (s, 6H, pyrrole –CH3), 2.08 (d, 4J(H,H) = 2.0 Hz, 2H, –CCH), 1.43 (s, 6H, pyrrole –CH3) ppm. 13C NMR (125.8 MHz, CDCl3, 293 K): δ 159.0, 155.3, 143.1, 141.7, 131.8, 129.3 (phenyl CH), 127.5, 121.1 (pyrrole CH), 115.2 (phenyl CH), 80.2 (–CCH), 70.1 (–CCH), 66.0 (–OCH2–), 19.6 (CH2CCH), 14.6 (both pyrrole –CH3 groups, signals overlap) ppm. 19F NMR (235.4 MHz, CDCl3, 293 K): δ −146.3 (q, 1J(F,B) = 33.2 Hz, BF2) ppm. HRMS (ESI): calc. [M + H]+: 393.1872; found 393.1919. MS (EI+ mode): calc. [M]+: 392.2; found 392.3. Calc. [M − HF]+: 372.2; found 372.3. IR: 3283, 2957, 2943, 2926, 2910, 2887, 1607, 1541, 1506, 1456, 1433, 1408, 1372, 1364, 1340, 1306, 1283, 1248, 1109, 1022, 1013, 892, 581 cm−1. MP: 151–154 °C (decomposition).
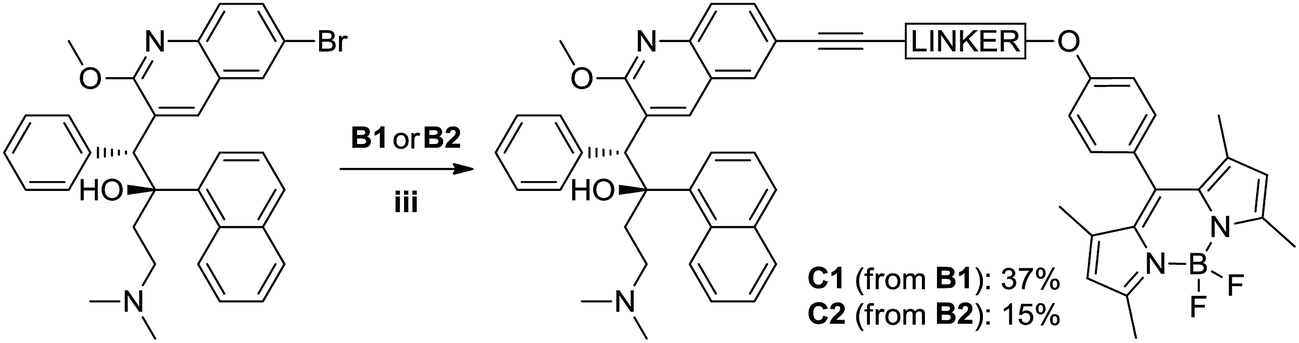
Synthesis of bedaquiline-butynyloxy-BODIPY (C1)
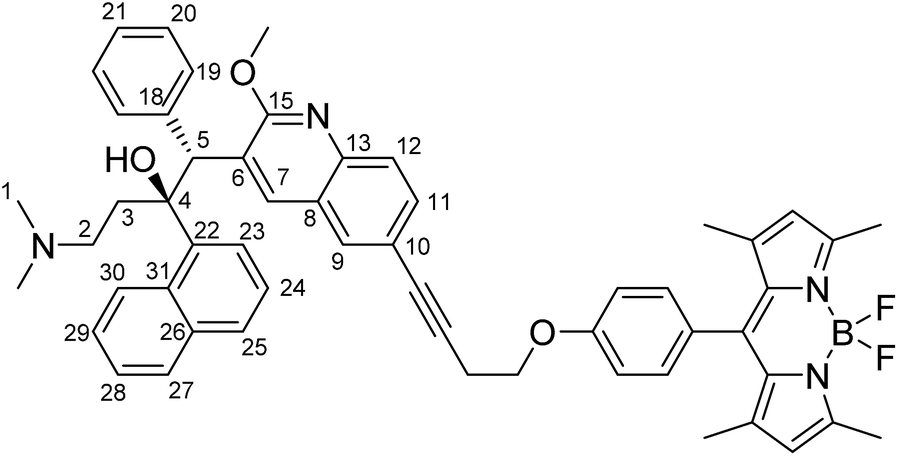
To a 20 mL glass reactor was added bedaquiline fumarate (68 mg, 102 μmol, 1.0 eq.) and butynoxy-BODIPY B1 (60 mg, 153 μmol, 1.5 eq.) in NEt3 (3 mL). The solution was degassed with argon for 5 minutes, after which CuI (2 mg, 10 μmol, 0.1 eq.) and Pd(PPh3)4 (12 mg, 10 μmol, 0.1 eq.) in 3 mL argon-degassed DMF was added via syringe from stock solutions prepared and kept under argon. The reactor was capped with a Teflon-lined plug. After stirring the reaction mixture at 90 °C for 2 hours, the reaction mixture was poured into 1:1 ethyl acetate:satd. NaHCO3. The ethyl acetate layer was washed twice with water and once with brine. The organic phase was dried over Na2SO4 and concentrated in vacuo. Column chromatography (first column SiO2 stationary phase using 6:1 pentane:EtOAc to 1:1 pentane:EtOAc; second column SiO2 stationary phase using 1:1 pentane:EtOAc to 100% EtOAc) afforded bedaquiline-butynyloxy-BODIPY C1 (Fig. 2, 32.5 mg, 38 μmol, 37%) as a bright orange solid.
TLC (Pentane:EtOAc 1:1): Rf = 0.4, red spot with green fluorescence. 1H NMR (500.2 MHz, CDCl3, 293 K): δ 8.94 (s, 1H, CH7), 8.61 (d, 1H, 3J(H,H) = 8.5 Hz, naphthyl CH), 7.94 (s, 1H, CH9), 7.89 (t, 2H, 3J(H,H) = 6.5 Hz, naphthyl CH), 7.77 (d, 1H, 3J(H,H) = 8.5 Hz, CH12), 7.66 (d, 1H, 3J(H,H) = 8.0 Hz, naphthyl CH), 7.62 (m, 1H, naphthyl CH; signals overlap), 7.61 (m, 1H, CH11; signals overlap), 7.49 (t, 1H, 3J(H,H) = 7.0 Hz, naphthyl CH), 7.31 (t, 1H, 3J(H,H) = 7.5 Hz, naphthyl CH), 7.20 (d, 2H, 3J(H,H) = 8.5 Hz, phenol CH), 7.12 (br. s, 2H, phenyl CH19), 7.08 (d, 2H, 3J(H,H) = 8.5 Hz, phenol CH), 6.89 (br. s, 3H, phenyl CH20/CH21), 5.98 (s, 2H, pyrrole CH), 5.89 (s, 1H, CH5), 4.28 (t, 2H, 3J(H,H) = 7.0 Hz, OCH2–), 4.22 (s, 3H, OCH3), 3.01 (t, 2H, 3J(H,H) = 7.0 Hz, –OCH2CH2–), 2.56 (s, 6H, pyrrole –CH3), 2.5 (1H, –CH23–, signals overlap), 2.13 (t, 1H, 3J(H,H) = 13 Hz, –CH23–), 1.98–1.92 (m, 2H, –CH22–; signals overlap), 1.94 (s, 6H, N(CH3)2), 1.45 (s, 6H, pyrrole –CH3) ppm. 13C NMR (125.8 MHz, CDCl3, 293 K): δ 161.6, 159.1, 155.3, 144.6, 143.2, 141.8, 140.6, 139.4, 134.7, 131.8, 131.8, 131.8, 131.3, 129.9, 129.9, 129.3, 128.2, 127.4, 127.3, 127.1, 126.8, 125.8, 125.3, 125.3, 125.2, 125.1, 124.5, 121.2, 118.5, 115.3, 85.3 (–CH2CC–), 82.3 (–CH2CC–), 66.4 (–OCH2–), 56.4 (C5), 54.2 (–OCH3), 49.5, 44.7 (N(CH3)2), 33.4, 20.6 (–CH2CC–), 14.7 (pyrrole CH3), 14.6 (pyrrole CH3) ppm. 19F NMR (235.4 MHz, CDCl3, 293 K): δ −146.3 (q, 1J(F,B) = 32.4 Hz, BF2) ppm. HRMS (ESI): calc. [M + H]+: 867.4252; found 867.4201. No bedaquiline starting material was found. IR: broad peak at 3000–2700 cm−1. 1543, 1508, 1454, 1400, 1369, 1350, 1306, 1286, 1242, 1192, 1155, 1119, 1080, 1038, 1014, 926, 833, 806, 737, 704 cm−1. MP: 137–145 °C (decomposition).
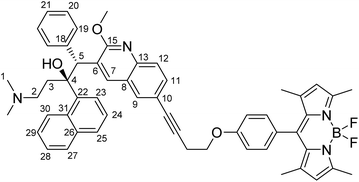 |
| | Fig. 2 Structural formula of bedaquiline-butynoxy-BODIPY. The numbering scheme used in this work for elucidation of NMR spectroscopic data is shown. | |
Synthesis of pentynyloxy-eg4-OH
To dry DMF (12 mL) and tetraethylene glycol mono THP ether (4.90 g, 17.6 mmol, 1.0 eq.), prepared according to Baker et al.18 and kept in a dry Schlenk flask under nitrogen atmosphere, was added NaOMe (1.19 g, 22.0 mmol, 1.25 eq.) at 0 °C. The clear solution was warmed up to room temperature over the course of 30 minutes. After again cooling down the reaction mixture to 0 °C, 5-chloro-1-pentyne (1.81 g, 17.6 mmol, 1.0 eq.) and of sodium iodide (0.15 g, 1 mmol, 5 mol%) were added. The reaction mixture was heated to 80 °C for 16 hours. Subsequently, the reaction mixture was quenched with a little aqueous saturated NaHCO3 solution, diluted with approximately 100 mL CH2Cl2 and extracted three times with water. The organic phase was concentrated in vacuo and dissolved in methanol (100 mL) to which was added para-toluenesulfonic acid monohydrate (150 mg). The methanolic solution was stirred at room temperature for 72 hours, after which it was concentrated in vacuo, dissolved in 100 mL CH2Cl2 and extracted once with saturated aqueous NaHCO3 and twice with water. The organic phase was dried over magnesium sulfate, concentrated in vacuo and subjected to column chromatography (SiO2 stationary phase, 100% EtOAc) to afford pentynyloxy-eg4-OH (695 mg, 2.67 mmol, 15%) as a colorless oil. TLC (100% EtOAc): Rf = 0.20, visualized using iodine vapour or CerMop staining. 1H NMR (500 MHz, CDCl3): δ 3.68 (2H, br. s), 3.6–3.5 (10H, m), 3.56 (4H, m), 3.51 (2H, t, 3J(H,H) = 6.3 Hz), 2.24 (2H, dt, 3J(H,H) = 7 Hz, 4J(H,H) = 2.5 Hz, HCCCH2–), 1.91 (1H, t, 3J(H,H) = 2.5 Hz, HCC–), 1.75 (2H, m, –CH2CH2CH2O–) ppm. 13C (125 MHz, CDCl3): δ 84.0 (–CCH), 72.6 (–CCH), 70.6, 70.6, 70.6, 70.3, 70.2, 69.6, 68.5, 61.7 (–OCH2CH2CH2–), 28.5 (–OCH2CH2CH2–), 15.2 (–OCH2CH2CH2–) ppm. HRMS (ESI): calc. [M + H]+: 261.1690 m/z. Found: 261.1697 m/z. Calc [M + Na]+: 283.1518 m/z. Found: 283.1516 m/z. IR: ν 3485.1, 3427.3, 2908.5, 2358.8, 1249.8, 1120.6, 1110.9, 1072.4, 1033.8, 731.0, 530.4, 505.3, 493.7m 418.5, 405.0 cm−1. Boiling point: 374 °C.
Synthesis of pentynyloxy-eg4-benzaldehyde (A2)
To dry THF (20 mL) kept at 0 °C under an N2 atmosphere was added pentynoxy-eg4-OH (550 mg, 2.11 mmol, 1.0 eq.), triphenylphosphine (583 mg, 2.11 mmol, 1.0 eq.) and 4-hydroxybenzaldehyde (257 mg, 2.11 mmol, 1.0 eq.). After five minutes of stirring, DEAD (918 mg, 2.11 mmol, 1.0 eq.) was added as a 40 wt% solution in toluene via a syringe. The solution was warmed up to room temperature over night; a color change from yellow to colorless was observed in the first 15 minutes. After 48 h, the solution was concentrated in vacuo. Column chromatography (SiO2 stationary phase, gradient elution 100% cyclohexane to 1:1 EtOAc:cyclohexane followed by a second SiO2 column using 8:1 EtOAc:CH2Cl2) afforded pentynyloxy-eg4-benzaldehyde A2 (259 mg, 0.71 mmol, 34%) as a colorless oil. TLC (1:1 EtOAc:cyclohexane): Rf = 0.25, visualized using 254 nm light. 1H NMR (400 MHz, CDCl3): δ 9.83 (s, 1H, –CHO), 7.78 (d, 1H, 3J(H,H) = 8.8 Hz, CH), 6.97 (d, 1H, 3J(H,H) = 8.8 Hz, CH), 4.16 (t, 2H, 3J(H,H) = 4.8 Hz, benzaldehyde-OCH2–), 3.84 (t, 2H, 3J(H,H) = 4.8 Hz, benzaldehyde-OCH2CH2–), 3.7–3.6 (m, 12H, –(OCH2CH2)3), 3.57 (t, 2H, 3J(H,H) = 6.3 Hz, –OCH2CH2CH2CCH), 2.23 (dt, 2H, 3J(H,H) = 6.9 Hz, 4J(H,H) = 2.8 Hz, HCCCH2), 1.91 (t, 1H, 4J(H,H) = 2.8 Hz, HCC–), 1.74 (m, 2H, –CH2CH2CH2O–) ppm. 13C NMR (100 MHz, CDCl3): δ 190.9 (–CHO), 163.9 (C1), 132.0 (C3), 130.00 (C4), 114.9 (C2), 84.0 (–CCH), 70.9, 70.6, 70.6, 70.6, 70.6, 70.2, 69.5, 69.4, 67.8, 63.6 (–OCH2CH2CH2–), 28.5 (–OCH2CH2CH2–), 15.2 (–OCH2CH2CH2–) ppm. HRMS (ESI): calc. [M + H]+: 365.1951 m/z. Found: 365.1959 m/z. Calc [M + Na]+: 387.1784 m/z. Found: 387.1778 m/z. IR: ν 2869.9, 1685.7, 1600.8, 1577.7, 1311.5, 1258.5, 1217.0, 1161.1, 1057.0, 833.2, 651.9 cm−1. Boiling point: 337 °C (decomposition).
Sonogashira test reaction forming 3-(quinolin-6-yl)prop-2-yn-1-ol
In a 10 mL glass reactor, 6-bromoquinoline (210.5 mg, 1.01 mmol, 1 eq.), 3-butyn-1-ol (141.0 mg, 2.01 mmol, 2 eq.), Pd(PPh3)4 (61.0 mg, 0.05 mmol, 5 mol%), CuBr (9.1 mg, 0.06 mmol, 6 mol%) and triethylamine (4 mL, 0.25 M) were dissolved, degassed using N2 and reacted by heating the mixture to 90 °C for 70 minutes.20 The reaction mixture was partitioned between a saturated aqueous solution of (NH4)2SO4 and diethyl ether. The water layer was made alkaline by addition of a saturated aqueous solution of NaHCO3 followed by extraction with diethyl ether. The combined organic layers were dried (Na2SO4), volatiles were removed in vacuo and the crude was subjected to column chromatography (SiO2, gradient of pure cyclohexane to ethyl acetate) to yield the product, which was obtained as an oil (151 mg, 0.78 mmol, 77%). TLC (2:1 cyclohexane:EtOAc): Rf = 0.50. 1H NMR (500 MHz, CDCl3): δ 8.89 (m, 1H), 8.08 (d, 1H, 3J(H,H) = 8.5 Hz), 8.01 (d, 1H, 3J(H,H) = 8.5 Hz), 7.88 (s, 1H), 7.60 (d, 1H, 3J(H,H) = 8.5 Hz), 7.40 (q, 1H, 3J(H,H) = 4 Hz), 3.87 (t, 2H, 3J(H,H) = 6.0 Hz), 2.75 (t, 2H, 3J(H,H) = 6.0 Hz), 2.28 (br, 1H, OH). 13C NMR (125 MHz, CDCl3): δ 150.9, 147.6, 135.8, 132.5, 131.2, 129.6, 128.1, 121.9, 121.8, 88.2, 82.1, 61.224.1.
Synthesis of pentynyloxy-eg4-BODIPY (B2)
To dry THF (30 mL) under an N2 atmosphere was added aldehyde A2 (200 mg, 0.55 mmol, 1.0 eq.), 2,4-dimethylpyrrole (115 mg, 1.21 mmol, 2.2 eq.) and 1 drop of trifluoroacetic acid. After 16 hours of stirring at room temperature, DDQ (125 mg, 0.55 mmol, 1.0 eq.) was added as a solution in 1 mL THF. After 1 hour of stirring, the reaction mixture was cooled to 0 °C using ice, and NEt3 (444 mg, 4.4 mmol, 8 eq.) of NEt3 was added via syringe followed by BF3·OEt2 (780 mg, 5.5 mmol, 10 eq.). The reaction mixture was allowed to warm up to room temperature over the course of two hours, after which it was filtered over celite. The celite was washed with CH2Cl2; pooled organic phases are concentrated in vacuo. The brown, sticky solid thus obtained was diluted with CH2Cl2 and washed with satd. NaHCO3, satd. NH4Cl, water and brine. The pooled aqueous layers were back-extracted once with CH2Cl2. After drying over Na2SO4, the organic layer was concentrated in vacuo. Column chromatography (SiO2 stationary phase, isocratic elution with 1:1 EtOAc:cyclohexane) gave pentynyloxy-eg4-BODIPY B2 (141.7 mg, 0.23 mmol, 54%) as a brown-orange waxy solid. TLC (1:1 EtOAc:cyclohexane): Rf = 0.20, red spot with green fluorescence. 1H NMR (500 MHz, CDCl3): δ 7.14 (d, 2H, 3J(H,H) = 8.5 Hz, phenol CH), 7.01 (d, 3J(H,H) = 8.5 Hz, 2H, phenol CH), 5.97 (s, 2H, pyrrole CH), 4.17 (t, 2H, 3J(H,H) = 5.0 Hz, –CH2–), 3.90 (t, 2H, 3J(H,H) = 5.0 Hz, –CH2–), 3.76–3.59 (m, 12H, –OCH2CH2O–), 3.55 (t, 2H, 3J(H,H) = 6.3 Hz, –OCH2(CH2)2CCH), 2.54 (s, 6H, pyrrole CH3), 2.28 (dt, 2H, –CH2CCH), 1.93 (t, 1H, –CCH), 1.79 (m, 2H, –OCH2CH2CH2–), 1.41 (s, 6H, pyrrole) ppm. 13C NMR (125 MHz, CDCl3): δ 159.4, 155.3, 143.3, 141.9, 132.0, 129.3 (phenol CH), 127.3, 121.2 (pyrrole CH), 115.3 (phenol CH), 84.1 (–CCH), 71.0, 70.8, 70.7, 70.4, 69.8, 69.7, 68.6, 67.6, 28.6 (–OCH2CH2CH2–), 15.3 (–CH2CCH), 14.8 (both pyrrole CH3 groups, signals overlap) ppm. 19F NMR (235.4 MHz, CDCl3, 293 K): δ −146.3 (q, 1J(F,B) = 32.9 Hz, BF2) ppm. HRMS (ESI): calc. [M + Na]+: 605.2969 m/z. Found: 605.2982 m/z. IR: ν 2922.0, 2864.1, 2358.8, 2337.6, 1608.5, 1541.0, 1508.2, 1467.7, 1407.9, 1363.6, 1305.7, 1286.4, 1245.9, 1191.9, 1155.3, 1982.0, 1049.2, 1012.6, 974.0, 853.1, 763.8, 704.0, 601.8, 582.5 cm−1. Melting point: 158–164 °C (decomposition).
Synthesis of bedaquiline-pentynoxy-eg4-BODIPY (C2)
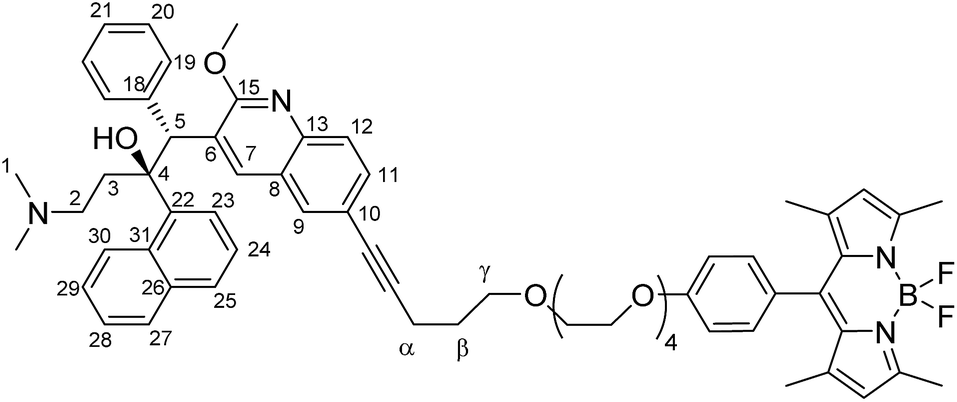
To a 10 mL glass reactor was added bedaquiline fumarate (76.6 mg, 114.5 μmol, 1.0 eq.) and pentynoxy-eg4-BODIPY B2 (99.7 mg, 172 μmol, 1.5 eq.) in 5 mL NEt3. The solution was degassed with argon for 5 minutes, after which CuI (1.1 mg, 6 μmol, 5 mol%) and Pd(PPh3)4 (26.4 mg, 23 μmol, 0.2 eq.) in 5 mL argon-degassed DMF were added via syringe from stock solutions prepared and kept under argon. The reactor was capped with a Teflon-lined plug. After stirring the reaction mixture at 90 °C for 2 hours, the reaction mixture was poured into 1:1 ethyl acetate:satd. NaHCO3. The ethyl acetate layer was washed twice with water and once with brine. The organic phase was dried over Na2SO4 and concentrated in vacuo. Extensive column chromatography (SiO2 stationary phase using 3:1 pentane:EtOAc to 1:1 pentane:EtOAc; followed by Al2O3 stationary phase using 8:1 hexane:EtOAc to 100% EtOAc) followed by washing of the obtained solid with about 5 mL of isopropanol gave bedaquiline-pentynoxy-eg4-BODIPY C2 (Fig. 3, 17.9 mg, 17.0 μmol, 15%) as a bright orange solid.
TLC (3:1 EtOAc:cyclohexane): Rf = 0.22, red spot, green fluorescence. 1H NMR (500 MHz, CDCl3): δ 8.86 (s, 1H), 8.54 (d, 1H, 3J(H,H) = 9.0 Hz), 8.31 (br. s, 1H, –OH), 7.81 (m, 3H), 7.67 (d, 1H, 3J(H,H) = 8.5 Hz), 7.58 (d, 1H, 3J(H,H) = 8.0 Hz), 7.54 (t, 1H, 3J(H,H) = 7.75 Hz), 7.50 (d, 1H, 3J(H,H) = 8.5 Hz), 7.42 (t, 1H, 3J(H,H) = 7.5 Hz), 7.23 (t, 1H, 3J(H,H) = 7.75 Hz), 7.08 (d, 2H, 3J(H,H) = 9.0 Hz, phenol CH), 6.95 (br. s, 2H), 6.94 (d, 2H, 3J(H,H) = 8.5 Hz, phenol CH), 6.81 (t, 3H, 3J(H,H) = 3 Hz), 5.98 (s, 2H, pyrrole CH), 5.82 (s, 1H, CH5), 4.13 (s, 3H, –OMe), 4.10 (t, 2H, 3J(H,H) = 4.6 Hz), 3.83 (t, 2H, 3J(H,H) = 5 Hz, –CH2–), 3.7–3.5 (m, 14H, –CH2–), 2.49 (s, 6H, pyrrole CH3), 2.5 (m, 2H, –CH2α–), 2.5 (m, 1H, diastereotopic –CH23– protons), 2.05 (t, 1H, 3J(H,H) = 12.5 Hz, diastereotopic –CH23– protons), 1.9 (m, 2H, –CH22–), 1.90 (s, 6H, –NMe2), 1.9–1.8 (m, 2H, –CHβ2–), 1.34 (s, 6H, pyrrole CH3) ppm. 13C NMR (125 MHz, CDCl3): δ 161.6, 159.5, 155.3, 144.5, 143.3, 142.0, 141.9, 140.8, 139.4, 134.8, 132.0, 131.1, 130.1, 130.0, 129.3 (phenol CH), 128.3, 127.5, 127.3, 127.2, 126.9, 125.5, 125.4, 125.4, 124.6, 121.2 (pyrrole CH), 119.3, 115.3 (phenol CH), 89.5, 82.8, 81.1, 71.0, 70.8, 70.5, 70.0, 69.9, 67.6, 56.5 (C2), 54.3 (OMe), 49.6 (C5), 44.9 (C1), 33.5 (C3), 29.0 (Cβ), 16.4 (Cα), 14.8 (pyrrole CH3), 14.7 (pyrrole CH3) ppm. 19F NMR (235.4 MHz, CDCl3, 293 K): δ −146.3 (q, 1J(F,B) = 32.9 Hz, BF2) ppm. HRMS (ESI): calc [M + H]+: 1057.5457, [M + H + Na]2+: 540.2675, [M + 2H]2+: 529.2765, [M + H–F]2+: 519.2734 m/z. Found resp. 1057.5426, 540.2656, 529.2739, 519.2715 m/z. IR: ν 2922.0, 2860.2, 2358.8, 2331.8, 1606.6, 1543.0, 1508.2, 1456.2, 1438.8, 1400.2, 1305.7, 1245.9, 1193.9, 1155.3, 1116.7, 1105.1, 1080.1, 1062.7, 1051.1, 1012.6, 975.9, 910.3, 835.1, 729.0, 704.0, 640.3, 603.7, 582.5, 455.2 cm−1 MP: 165–170 °C (decomposition).
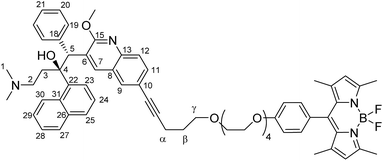 |
| | Fig. 3 Structural formula of bedaquiline-pentynoxy-eg4-BODIPY C2. The numbering scheme used in this work for elucidation of NMR spectroscopic data is shown. | |
Results and discussion
Synthesis of fluorescent alkynyl-BODIPY labels
Considering the presence of an aryl bromide moiety in the molecular structure of bedaquiline, we arrived at a labeling strategy using Sonogashira cross-coupling chemistry to form a carbon–carbon bond between the bromoquinoline position and an alkyne-functionalized fluorophore.15 In principle, labeling of bedaquiline on the 6-position of the quinoline ring is expected to keep binding affinities towards the microbial target protein intact, as modification of the 6-position of the quinoline moiety has been previously applied for covalent linkage to a Biacore chip and an affinity resin.6 Furthermore, the bromoquinoline moiety has been shown to project away from the central binding pocket in the c-subunit of ATP synthase, with the bromo-moiety located at the distal position.14 Notwithstanding the apparent feasibility of this approach, it is obvious that close proximity of the fluorophore will lead to undesired fluorophore–protein interactions. Thus, we chose to investigate the synthesis of two alkyne-functionalized BODIPY13 fluorophores, which were subsequently used to prepare two fluorescently labeled bedaquiline analogues having a shorter (∼6 Å) or longer (∼22 Å) linear spacer between drug and fluorophore.
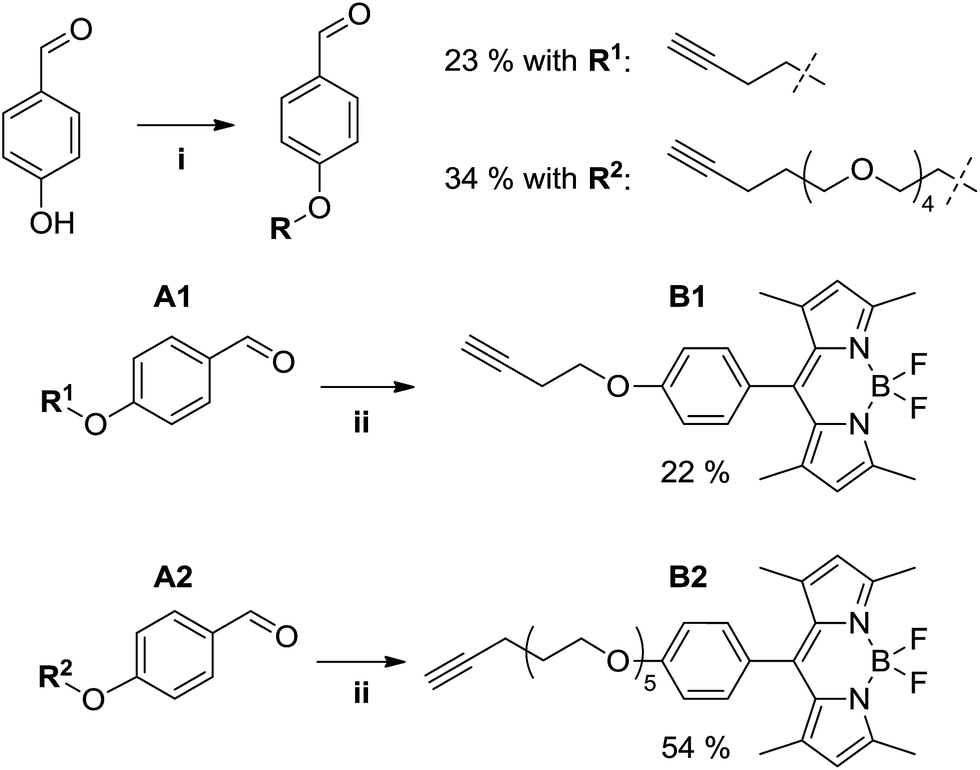
Fluorescent alkynyl-BODIPY derivatives B1 and B2 were prepared using a two-step route (see Fig. 4) starting from the Mitsunobu aryl ether synthesis of aldehydes A1 and A2, where the phenolic oxygen of 4-hydroxybenzaldehyde is alkylated by a butynyloxy or pentynyloxy-tetraethyleneglycol terminal alkyne, respectively. Subsequent acid-catalyzed condensation of the benzaldehyde with 2,4-dimethylpyrrole followed by oxidation and boron trifluoride chelation allowed us to obtain the target fluorophores, albeit in rather poor overall yields of 5% (B1) and 18% (B2) due to the typically poor yields of BODIPY preparations,13 and the difficulties encountered in separating the aryl ethers from the byproducts of the Mitsunobu reaction.
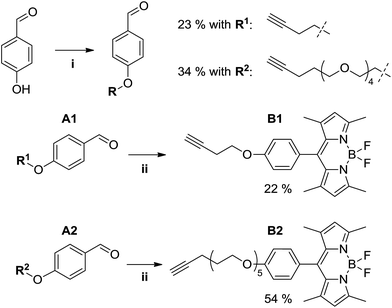 |
| | Fig. 4 Synthesis of alkyne-functionalized BODIPY derivatives. Reaction conditions: (i) 4-hydroxybenzaldehyde (1 eq.), alcohol R1-OH or R2-OH (1 eq.), PPh3 (1.05 eq.), DEAD (40 wt% in toluene, 1 eq.), THF, 0 °C → rt, overnight. (ii) A (1 eq.), 2,4-dimethylpyrrole (2.2 eq.), TFA (cat.), THF, rt, 16 h; then DDQ (1 eq.), rt, 1 h; then NEt3 (excess), BF3·OEt2 (excess), 0 °C → rt, 2 h. | |
Sonogashira-based labeling of bedaquiline
Initially, we ascertained the synthetic feasibility of our labeling strategy by reacting 3-butyn-1-ol with 6-bromoquinoline as model for bedaquiline under typical18 Sonogashira conditions (1.2:1.0 Cu:Pd stoichiometry), affording the desired coupling product in 77% isolated yield. We found that a direct translation of these conditions to the reaction of our BODIPY labels with bedaquiline led to the formation of the labeled drug, contaminated with a significant amount of the homo-coupled diyne as a side product. We speculate that the presence of the bedaquiline moiety is to blame for the appearance of this side product through the action of its Lewis basic sites, which can serve as a competitive bidentate ligand to one of the catalytically active metal species.19,21 However, encouraged by the presence of the target molecule, we then attempted to prepare the labeled bedaquiline analogues using relatively little CuI catalyst with respect to Pd0 (1:4 Cu:Pd stoichiometry); considering that the homocoupled bisalkyne is likely formed via the copper-catalysed Glaser coupling mechanism,22 lowering the relative copper concentration should suppress this side reaction.23 Under these conditions, a satisfactory conversion to the target compound was observed. Repeated purification was required to remove any traces of the unreacted parent drug, which accounts for the poor yields of, respectively, 37% and 15% for C1 and C2 (see Fig. 5).
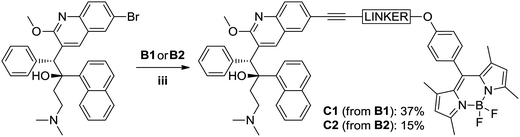 |
| | Fig. 5 Labeling of bedaquiline using a Sonogashira coupling. Reaction conditions. (iii) Bedaquiline fumarate 1 eq., Bodipy 1.5 eq., CuI 0.05 eq., Pd(PPh3)4 0.2 eq. in 10 mL 1:1 DMF:NEt3 added under Argon atmosphere at rt; reaction heated at 90 °C for 2 h. | |
Photophysical properties of fluorescent bedaquiline analogues
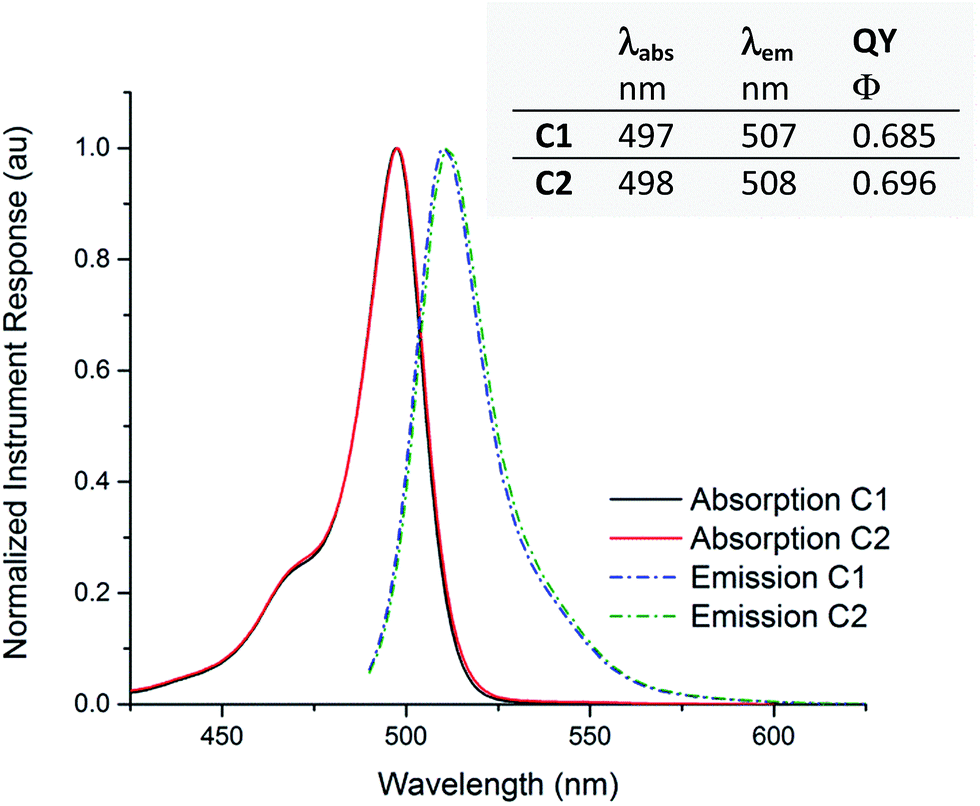
Using steady-state light absorption and fluorescence measurements, acetonitrile solutions of compounds C1 and C2 were characterized photophysically (see Fig. 6). In the chosen solvent, varying distance between label and drug appears to have no influence on the properties under study, as virtually identical wavelength maxima λabs and λem were observed, with a Stokes shift of 10 nm for both compounds. The quantum yield of fluorescence is also independent of linker identity, showing the expected isolation of the fluorophore from linker and drug. The absorption and emission wavelength maxima of C1 and C2 are essentially identical in methanol, a protic solvent of similar polarity to acetonitrile (see ESI†). We also confirmed that the parent compound bedaquiline did not show emission peaks in the 400–600 nm wavelength range (not shown in Fig. 6). As is typical for BODIPY derivatives13 no changes in the absorption spectrum were seen when a cuvette of C1 was left standing under ambient illumination for several weeks; nor could we over time observe any degradation in the 1H NMR spectrum of C1. The labeled bedaquiline derivatives described can thus be regarded as stable, suitable fluorescent probes for biological applications.
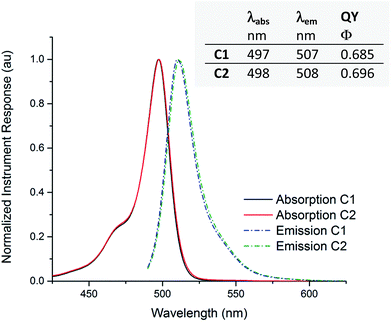 |
| | Fig. 6 Emission and absorption curves for compounds C1 and C2. Normalized absorption (solid lines) and emission (dashed lines) plots, recorded in acetonitrile solution. The inset tabulates absorption and emission wavelength maxima λabs and λem and the corresponding quantum yield of fluorescence Φ, calculated using fluorescein in 0.1 M aqueous NaOH as a reference. Peak normalized instrument responses for absorption measurements correspond to an extinction coefficient of respectively ε = 7.6 × 104 (C1) or 6.8 × 104 (C2) cm−1 M−1. | |
BODIPY-labeled bedaquiline compounds inhibit ATP synthesis
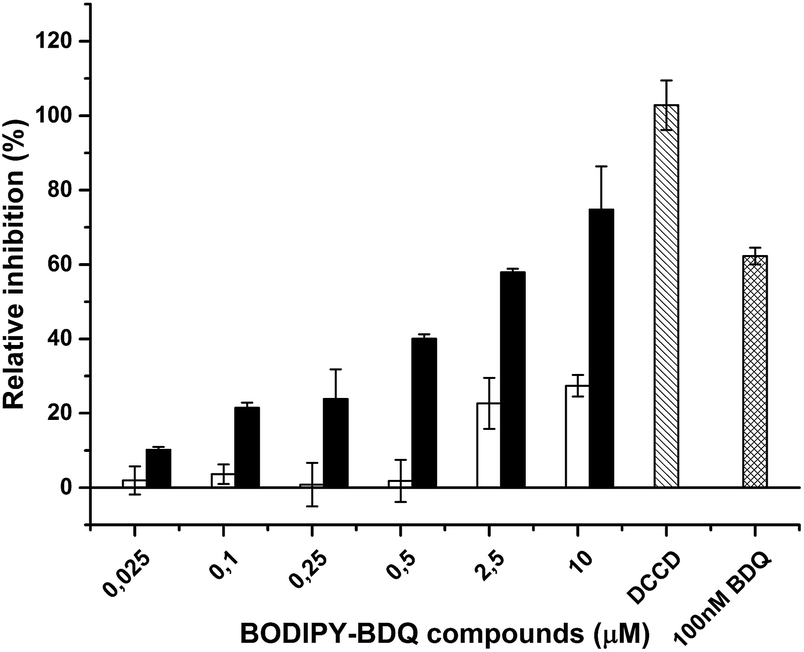
After successful preparation of the BODIPY-bedaquiline compounds, we sought to ascertain their antibacterial action, and to investigate if their mode of action is sufficiently similar to the parent compound; only sufficiently parent-like probe molecules are expected to yield useful results in follow-up studies on the mode of action of diarylquinoline antibiotics. To do so, the ability of C1 and C2 to inhibit the function of the biological target of the parent drug, ATP synthase, was evaluated. We measured the impact of the compounds on ATP synthesis activity using inverted membrane vesicles derived from Mycobacterium smegmatis. As depicted in Fig. 7, both C1 and C2 blocked the ATP synthesis activity in a dose-dependent manner. However, inhibition was clearly more efficient in case of C2 (IC50 ∼ 2.0 μM). The lower activity of C1 (IC50 > 10 μM) may be due to the shorter linker, interfering with effective binding of the compound to its binding pocket, which is located close to the center of the hydrophobic, membrane-spanning part of ATP synthase.16 These IC50 values are higher than those reported for the parent compound (IC50 between 10–100 nM),7,24 but comparable to and consistent with values reported for fluorescent derivatives of other anti-tuberculosis drugs, e.g. for benzothiadizinones.13 The results conclusively prove that the presented labeling strategy allows the preparation of bedaquiline analogues that retain the ability to inhibit ATP synthesis, given the correct spacing distance between label and drug.
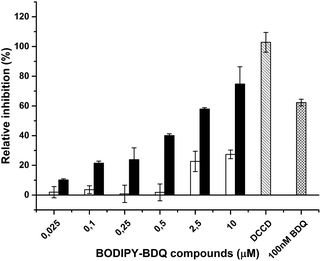 |
| | Fig. 7 Inhibition of mycobacterial ATP synthesis activity by BODIPY-bedaquiline (BDQ) compounds. ATP synthesis activity was measured in inverted membrane vesicles from M. smegmatis using the glucose-6-phosphate dehydrogenase method. BODIPY-bedaquiline compounds (open bars: C1, butynyloxy linker, closed bars, C2, pentynyloxy-eg4 linker) were added at the indicated concentrations. 100 nM BDQ and the ATP synthase inhibitor DCCD were used as control. | |
BODIPY-labeled bedaquiline compounds inhibit bacterial growth
Next, we evaluated their ability to interfere with growth of living bacteria. As shown in Table 1, both C1 and C2 suppressed growth of M. tuberculosis, with minimal inhibitory concentrations of 1.8 μM and 7.3 μM, respectively (Table 1), compared to 0.1 μM for the parent compound bedaquiline.6 This indicates that labeling of bedaquiline with the BODIPY moiety and introduction of the linker decreased, but did not abolish, the antibacterial activity of the drug. In M. tuberculosis, efflux pumps can be an important determinant for the sensitivity towards antibacterials.17 Recently it was reported that the efflux pump inhibitor verapamil enhanced the susceptibility of M. tuberculosis for bedaquiline17 whereas upregulation of the mycobacterial efflux pump MmpS5–MmpL5 decreased the sensitivity for this drug.14,17 Therefore we evaluated the metabolic similarity of the labeled compounds to bedaquiline by checking if they are also recognized by the efflux system. Addition of the efflux pump inhibitor verapamil increased the sensitivity of M. tuberculosis reference strains H37Rv for C1 and C2 (respectively twofold and eightfold, Table 1), suggesting that both BODIPY-bedaquiline analogues are indeed transported by an efflux pump. We also tested growth inhibition of two M. tuberculosis strains with elevated levels of the MmpS5–MmpL5 efflux pump: one strain carrying a mutation in the regulator of MmpS5–MmpL5 (CV37) and one laboratory strain carrying a plasmid for over-expression of MmpS5–MmpL5 (H37Rv pCVGA30). Both efflux pump overexpressing strains displayed considerably lower sensitivity towards C1 and C2, as well as to the parent compound (Table 1), suggesting that the fluorescent analogues are substrates of the same efflux system as bedaquiline.
Table 1 Growth inhibition of M. tuberculosis by BODIPY-bedaquilines. Strains tested were a M. tuberculosis reference strain (H37Rv) an in vitro generated Rv0678 mutant with increased efflux pump activity against BDQ (CV37), the reference strain with over-expressed efflux pumps (H37Rv pCVGA30) and with empty overexpression plasmid as control (H37Rv pSD5). VER: efflux pump inhibitor verapamil. BDQ = bedaquiline. Minimal inhibitory concentrations (MICs) were measured using the resazurin method as in ref. 14 and are expressed in mol L−1
| Strain |
BDQ |
BDQ + VER |
C1
|
C1 + VER |
C2
|
C2 + VER |
| H37Rv |
1.1 × 10−7 |
7.0 × 10−9 |
1.8 × 10−6 |
9.1 × 10−7 |
7.3 × 10−6 |
9.1 × 10−7 |
| CV37 |
9.1 × 10−7 |
2.3 × 10−7 |
>2.9 × 10−5 |
>2.9 × 10−5 |
>2.9 × 10−5 |
>2.9 × 10−5 |
| H37Rv pCVGA30 |
4.5 × 10−7 |
<5.7 × 10−8 |
7.3 × 10−6 |
9.1 × 10−7 |
>2.9 × 10−5 |
>2.9 × 10−5 |
| H37Rv pSD5 |
1.1 × 10−7 |
7.0 × 10−9 |
1.8 × 10−6 |
4.5 × 10−7 |
3.6 × 10−6 |
9.1 × 10−7 |
Conclusions
The obtained results show that we successfully designed a labeling strategy for bedaquiline that generates compounds sufficiently similar to the parent drug to be of use in scientific research. The obtained fluorescent analogues C1 and C2 can successfully interfere with ATP synthesis, the biological target of bedaquiline. Growth of living M. tuberculosis bacteria is suppressed at approximately 20- or 70-fold higher minimum inhibitory concentrations compared to bedaquiline, dependent on the spacing (6 Å versus 22 Å, respectively) between fluorescent label and drug. The probe compounds also share the affinity for the MmpS5–MmpL5 drug efflux system with the parent compound. Thus, fluorescent antibiotics prepared using the method described in this work are promising tools for deeper fluorescence-based experimentation on diarylquinoline-based tuberculosis drugs. Applications may include investigation of drug in/efflux at the level of either the bacterial population or the individual mycobacterial cell, determination of sub-cellular drug or target localization in the bacterium, as well as biochemical studies on binding of the drug to its target.
Acknowledgements
P. L. is indebted to a fellowship from the Chinese Scholarship council. J. A. R. was supported by the BioSolar Cells Programme of the Dutch Ministry of Economic Affairs. D. B. and J. C. S. thank AIMMS (Vrije Universiteit Amsterdam) for a stimulus grant (2011.A503). The authors wish to thank Peggy Janssens for technical assistance and W. Bas Wittekoek and Yvon van der Hulst for preliminary experiments.
Notes and references
-
World Health Organization, Global tuberculosis report 2015, World Health Organization, 2015 Search PubMed.
- A. Koul, E. Arnoult, N. Lounis, J. Guillemont and K. Andries, Nature, 2011, 469, 483–490 CrossRef CAS PubMed.
- D. Bald and A. Koul, FEMS Microbiol. Lett., 2010, 308, 1–7 CrossRef CAS PubMed; J. G. Hurdle, A. J. O'Neill, I. Chopra and R. E. E. Lee, Nat. Rev. Microbiol., 2011, 9, 62–75 CrossRef PubMed; P. A. Black, Antimicrob. Agents Chemother., 2014, 58, 2491–2503 CrossRef PubMed; P. Lu, H. Lill and D. Bald, Biochim. Biophys. Acta, 2014, 1837, 1208–1218 CrossRef PubMed; G. M. Cook, K. Hards, C. Vilcheze, T. Hartman and M. Berney, Microbiol. Spectrum, 2014, 2 Search PubMed.
- H. I. Boshoff, T. G. Myers, B. R. Copp, M. R. McNeil, M. A. Wilson and C. E. Barry, J. Biol. Chem., 2004, 279, 40174–40184 CrossRef CAS PubMed; E. A. Weinstein, T. Yano, L. S. Li, D. Avarbock, A. Avarbock, D. Helm, A. A. McColm, K. Duncan, J. T. Lonsdale and H. Rubin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4548–4553 CrossRef PubMed; T. Yano, S. Kassovska-Bratinova, J. S. Teh, J. Winkler, K. Sullivan, A. Isaacs, N. M. Schechter and H. Rubin, Biol. Chem., 2011, 286, 10276–10287 CrossRef PubMed.
- K. A. Abrahams, J. A. G. Cox, V. L. Spivey, N. J. Loman, M. J. Pallen, C. Constantinidou, R. Fernández, C. Alemparte, M. J. Remuiñán, D. Barros, L. Ballell and G. S. Besra, PLoS One, 2012, 7, e52951 Search PubMed; K. Pethe, P. Bifani, J. Jang, S. Kang, S. Park, S. Ahn, J. Jiricek, J. Jung, H. Kyoung Jeon, J. Cechetto, T. Christophe, H. Lee, M. Kempf, M. Jackson, A. J. Lenaerts, H. Pham, V. Jones, M. J. Seo, Y. M. Kim, M. Seo, J. J. Seo, D. Park, Y. Ko, I. Choi, R. Kim, S. Y. Kim, S. Lim, S. Yim, J. Nam, H. Kang, H. Kwon, C. Oh, Y. Cho, Y. Jang, J. Kim, A. Chua, B. Tan, M. B. Nanjundappa, S. P. S. Rao, W. S. Barnes, R. Wintjens, J. R. Walker, S. Alonso, S. Lee, J. Kim, S. Oh, T. Oh, U. Nehrbass, S. Han, Z. No, J. Lee, P. Brodin, S. Cho, K. Nam and J. Kim, Nat. Med., 2013, 19, 1157–1160 CrossRef CAS PubMed.
- K. Andries, P. Verhasselt, J. Guillemont, H. W. Göhlmann, J. M. Neefs, H. Winkler, J. Van Gestel, P. Timmerman, M. Zhu, E. Lee, P. Williams, D. de Chaffoy, E. Huitric, S. Hoffner, E. Cambau, C. Truffot-Pernot, N. Lounis and V. Jarlier, Science, 2005, 307, 223–227 CrossRef CAS PubMed; A. C. Haagsma, R. Abdillahi-Ibrahim, M. J. Wagner, K. Krab, K. Vergauwen, J. Guillemont, K. Andries, H. Lill, A. Koul and D. Bald, Antimicrob. Agents Chemother., 2009, 53, 1290–1292 CrossRef PubMed; A. C. Haagsma, I. Podasca, A. Koul, K. Andries, J. Guillemont, H. Lill and D. Bald, PLoS One, 2011, 6, e23575 Search PubMed; W. Balemans, L. Vranckx, N. Lounis, O. Pop, J. Guillemont, K. Vergauwen, S. Mol, R. Gilissen, M. Motte, D. Lançois, M. De Bolle, K. Bonroy, H. Lill, K. Andries, D. Bald and A. Koul, Antimicrob. Agents Chemother., 2012, 56, 4131–4139 CrossRef PubMed.
- A. Koul, N. Dendouga, K. Vergauwen, B. Molenberghs, L. Vranckx, R. Willebrords, Z. Ristic, H. Lill, I. Dorange, J. Guillemont, D. Bald and K. Andries, Nat. Chem. Biol., 2007, 3, 323–324 CrossRef CAS PubMed.
- A. Koul, L. Vranckx, N. Dhar, H. W. Göhlmann, E. Özdemir, J. M. Neefs, M. Schulz, P. Lu, E. Mørtz, J. D. McKinney, K. Andries and D. Bald, Nat. Commun., 2014, 5, 3369 Search PubMed; J. Dhillon, K. Andries, P. P. Phillips and D. A. Mitchison, Tuberculosis, 2010, 5, 301–305 CrossRef PubMed.
- A. H. Diacon, A. Pym and M. Grobusch, N. Engl. J. Med., 2009, 360, 2397–2405 CrossRef CAS PubMed; A. H. Diacon, Am. J. Respir. Crit. Care Med., 2015, 191, 943–953 CrossRef PubMed; S. Tyagi, N. C. Ammerman, S. Y. Li, J. Adamson, P. J. Converse, R. V. Swanson, D. V. Almeida and J. H. Grosset, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 869–874 CrossRef PubMed.
- J. Neres, F. Pojer, E. Molteni, L. R. Chiarelli, N. Dhar, S. Boy-Röttger, S. Buroni, E. Fullam, G. Degiacomi, A. P. Lucarelli, R. J. Read, G. Zanoni, D. E. Edmondson, E. De Rossi, M. R. Pasca, J. D. McKinney, P. J. Dyson, G. Riccardi, A. Mattevi, S. T. Cole and C. Binda, Sci. Transl. Med., 2012, 150, 150ra121 Search PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- P. Lu, M. H. Heineke, A. Koul, K. Andries, G. M. Cook, H. Lill, R. van Spanning and D. Bald, Sci. Rep., 2015, 5, 10333 CrossRef PubMed.
- P. Lu, P. A. C. Haagsma, H. Pham, J. J. Maaskant, S. Mol, H. Lill and D. Bald, Antimicrob. Agents Chemother., 2011, 55, 5354–5357 CrossRef CAS PubMed.
- K. Andries, C. Villellas, N. Coeck, K. Thys, T. Gevers, L. Vranckx, N. Lounis, B. C. de Jong and A. Koul, PLoS One, 2014, 9, e102135 Search PubMed.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- L. Preiss,
et al.
, Sci. Adv., 2015, 1, e1500106 Search PubMed; S. Singh, K. K. Roy, S. R. Khan, V. K. Kashyap, A. Sharma, S. Jaiswal, S. K. Sharma, M. Y. Krishnan, V. Chaturvedi, J. Lal, S. Sinha, A. Dasgupta, R. Srivastava and A. K. Saxena, Bioorg. Med. Chem., 2015, 4, 742–752 CrossRef PubMed.
- M. Viveiros, M. Martins, L. Rodrigues, D. Machado, I. Couto, J. Ainsa and L. Amaral, Expert Rev. Anti-Infect. Ther., 2012, 9, 983–998 CrossRef PubMed; K. N. Adams, J. D. Szumowski and L. Ramakrishnan, J. Infect. Dis., 2014, 3, 456–466 CrossRef PubMed; T. Coelho, D. Machado, I. Couto, R. Maschmann, D. Ramos, A. von Groll, M. L. Rossetti, P. A. Silva and M. Viveiros, Front Microbiol., 2015, 6, 330 Search PubMed; S. Gupta, K. A. Cohen, K. Winglee, M. Maiga, B. Diarra and W. R. Bishai, Antimicrob. Agents Chemother., 2014, 58, 574–576 CrossRef PubMed; R. C. Hartkoorn, S. Uplekar and S. T. Cole, Antimicrob. Agents Chemother., 2014, 58, 2979–2981 CrossRef PubMed.
- Q. Zhang, H. Ren and G. L. Baker, J. Org. Chem., 2014, 79, 9546–9555 CrossRef CAS PubMed.
- M. Meyer, S. Kuntz, I. Grillier-Vuissoz, H. Martin, L. Richert, S. Flament, Y. Chapleur and M. Boisbrun, Lett. Drug Des. Discovery, 2014, 11, 256–264 CrossRef CAS.
- L. Guandalini, E. Martini, S. Dei, D. Manetti, S. Scapecchi, E. Teodori, M. Romanelli, K. Varani, G. Greco, L. Spadola and E. Novellino, Bioorg. Med. Chem., 2005, 13, 799–807 CrossRef CAS PubMed.
- G. Chen, W. Gong, Z. Zhuang, M. S. Andra, Y. Chen, X. Hong, Y. Yang, T. Liu, K. N. Houk and J. Yu, Science, 2016, 6303, 1023–1027 CrossRef PubMed.
- I. D. Campbell and G. Eglinton, Org. Synth., 1965, 45, 39 CrossRef CAS.
- M. Karak, L. C. A. Barbosa and G. C. Hargaden, RSC Adv., 2014, 4, 53442–53466 RSC.
- Bedaquiline showed approx. 60% inhibition of ATP synthesis under our conditions at 100 nM, suggesting a 25-fold difference in activity between bedaquiline and C2.
Footnote |
| † Electronic supplementary information (ESI) available: In the supporting information, a pdf document is available containing all NMR spectroscopic data pertaining to the compounds mentioned, as well photochemical characterization of compounds C1 and C2. See DOI: 10.1039/c6ra22693k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Romano V. A.
Orru
b,
Koop
Lammertsma
ae,
Dirk
Bald
*d and
J. Chris
Slootweg
*a
b,
Romano V. A.
Orru
b,
Koop
Lammertsma
ae,
Dirk
Bald
*d and
J. Chris
Slootweg
*a