
Natural polysaccharides leading to super adsorbent hydroxyapatite nanoparticles for the removal of heavy metals and dyes from aqueous solutions†
Abstract
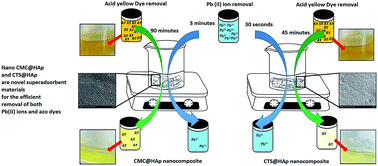
Water pollution has created a major impact on the environment mainly due to contaminated industrial effluents with toxic substances such as heavy metals and textile dyes. Therefore finding effective methodologies for their removal are extremely important. Hydroxyapatite (HAp) is a biocompatible material and has been widely used in many biological and industrial applications. This study involves the synthesis of porous HAp polymer nanocomposites using chitosan (CTS@HAp) and carboxymethyl cellulose (CMC@HAp) to develop a potential adsorbent with high adsorption capacity for heavy metals and textile dyes. A facile in situ synthetic approach was followed to obtain nanoparticles without calcination or freeze drying. X-ray diffraction, Fourier transform infra-red spectroscopy, scanning electron microscopy, thermogravimetric analysis, X-ray fluorescence spectroscopy and BET surface area characterization techniques were applied to elucidate the crystallinity, surface chemistry, morphology and surface area of HAp nanoparticles. This simple approach has produced 25–30 nm and 10 nm spherical nanoparticles of HAp in the presence of chitosan (CTS) and carboxymethyl cellulose (CMC) respectively. These nanocomposites were used for the removal of Pb(II) ions and an industrial dye waste, acid yellow 220. The equilibrium sorption data were fitted according to the Freundlich and Langmuir isotherm models. For the CMC@HAp system the equilibrium for adsorption was achieved in 3 min, while CTS@HAp system required only 30 seconds. The maximum Pb(II) ion adsorption capacity (qm) was found to be 625.0 mg g−1 and 909.1 mg g−1 for CMC@HAp and CTS@HAp respectively. In the case of acid yellow 220, the equilibrium for adsorption was achieved in 90 minutes and 45 minutes for CMC@HAp and CTS@HAp respectively. The maximum acid yellow 220 adsorption capacity (qm) was found to be 200 mg g−1 and 303 mg g−1 for CMC@HAp and CTS@HAp respectively. Therefore this study has highlighted the possibility of developing a HAp nanocomposite material for the efficient and effective removal of both Pb(II) ions and acid yellow 220 from aqueous system.
 Please wait while we load your content...
Please wait while we load your content...

