
Direct amide synthesis over core–shell TiO2@NiFe2O4 catalysts in a continuous flow radiofrequency-heated reactor
 a
and
a
and
Abstract
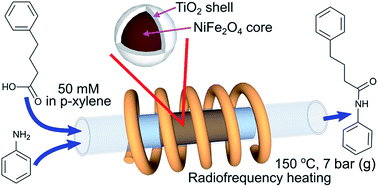
Core–shell composite magnetic catalysts TiO2@NiFe2O4 with a titania loading of 9–32 wt% have been synthesised by a sol–gel method for direct amide synthesis in a radiofrequency (RF, induction)-heated continuous flow reactor. The catalyst calcination temperature was optimised in the range of 350–500 °C and the highest activity was observed for the catalyst calcined at 500 °C due to conversion of titania into the catalytically active anatase phase. No reaction between the magnetic core and the titania shell was observed up to the calcination temperature of 1000 °C and no sintering of the titania shell was observed after calcination at 500 °C. The comparison of direct amide synthesis in a continuous flow fixed bed reactor under conventional and RF heating demonstrated that the RF heating mode increased the apparent reaction rate by 60% and decreased the deactivation rate due to a better temperature uniformity. The titania weight normalised reaction rate in the RF-heated reactor was constant for titania loadings above 17 wt%, while it decreased by a factor of 3 at lower titania loadings because of interactions between the ferrite core on the thin layer of the catalyst. The catalyst deactivation study showed that the deactivation rate could be accurately described by first order kinetics and that the main reason of deactivation was coking. The catalyst regeneration via calcination at 400 °C resulted in catalyst sintering, while treatment with a hydrogen peroxide solution at 90 °C fully recovered the catalytic activity.
 Please wait while we load your content...
Please wait while we load your content...

