
Membraneless flow battery leveraging flow-through heterogeneous porous media for improved power density and reduced crossover
Abstract
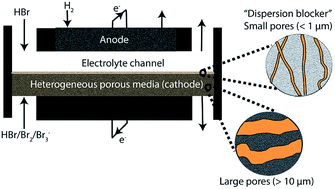
We propose and demonstrate a novel flow battery architecture that replaces traditional ion-exchange membranes with less expensive heterogeneous flow-through porous media. Compared to previous membraneless systems, our prototype exhibits significantly improved power density (0.925 W cm−2), maximum current density (3 A cm−2), and reactant crossover, shown by the proposed experimentally-validated crossover model.
 Please wait while we load your content...
Please wait while we load your content...

