DOI:
10.1039/C6RA22549G
(Paper)
RSC Adv., 2016,
6, 111366-111373
Iron nanoparticles with a square pyramidal structure in mesoporous carbons as an effective catalyst toward oxygen reduction†
Received
9th September 2016
, Accepted 16th November 2016
First published on 16th November 2016
Abstract
Mesoporous carbons supported well-dispersed square pyramidal structures of Fe–Nx (FeAMC-T) have been fabricated by pyrolyzing amino-functionalized ionic liquids and iron precursors in a mesoporous silica. The fabricated FeAMC-T is heat-treated in a nitrogen environment at various temperatures (873–1273 K) to obtain optimized catalysts for the oxygen reduction reaction (ORR). The electrochemical activities of the catalysts are investigated by rotating disk electrode tests in 0.5 M H2SO4 saturated with oxygen. A series of different spectroscopic (X-ray diffraction (XRD), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS)) and analytical techniques (N2 adsorption–desorption isotherms and elemental analysis (EA)) are used to characterize the mesoporous structure, morphology and chemical environment of the catalysts. The FeAMC-1273 catalysts, which are most likely to possess a high specific surface area (1002 m2 g−1) with moderate nitrogen doping (∼2.8 wt%) on mesoporous carbons and more pyridinic-N (34.9%) as well as pyridinic-N–Fe (52.3%) species for the creation of a square pyramidal planar geometry around iron (confirmed by XAS), exhibit a four-electron transfer process, the best ORR activity (onset potential = 0.69 V vs. Ag/AgCl) and methanol-resistant durability in acid solution while compared to the commercially available catalysts (20 wt% of Pt on activated carbon).
Introduction
Direct methanol fuel cells (DMFCs) have been regarded as one potential energy device due to their high conversion efficiency, low operational temperature and low pollution.1,2 To speed up the large-scale applications of DMFCs, efficient electrocatalysts with low costs for the oxygen reduction reaction (ORR) in the cathode are urgently needed. At present, platinum (Pt) and Pt-based alloys electrodes are the best ORR catalysts in terms of their highly electrocatalytic properties.3–11 Nevertheless, the high price and worse tolerance to the methanol crossover of Pt hinder their extensive commercialization.12–16 Therefore, in order to realize viable utilization of DMFCs, non-precious metal17–23 and non-metal catalysts24–28 have been widely investigated. In general, non-precious metal catalysts were achieved by heat-treating a transition metal (e.g., iron or cobalt) together with nitrogen-rich compounds (for example ethylenediamine, phthalocyanine, porphyrin, phenanthroline, aminobenzimidazole and tripyridyl triazine, etc.) in an inert environment.29–35 Among them, transition metal–macrocyclic catalysts (particularly metal phthalocyanine macrocycles)36–40 have been studied as a possible option to replace Pt and its alloys catalysts. Additionally, iron phthalocyanine complexes were shown to have superior ORR performance compared to that of other metal phthalocyanine complexes. Above all, it is reported41–44 that Fe–N4 structure (square planar structure) would be the active sites for ORR. Nonetheless, the ORR electrocatalytic activity of pentacoordinate iron phthalocyanine electrocatalysts (square pyramidal planar structure) is also shown to be superior to that of tetracoordinate iron phthalocyanine electrocatalysts (square planar structure).45 Thus, the molecular-scale structure of Fe–Nx species is a critical factor that can influence the ORR activity.
Recently, ionic liquids (ILs)46–50 are explored to be prospective precursors for the synthesis of N-doped carbons because of their distinctive properties, including insignificant vapor pressures, superior stability and adjustable solubility as well as flexibility for chemical synthesis. Moreover, mesoporous carbons possess tunable pore structure as well as high surface area which can increase the rate of mass transfer and also offer more exposure of active sites. In the recent years, the design of precursors at the dimension of molecular scale may be helpful to adjust the chelating capability between transition metals and nitrogen precursors.51,52 Upon synthesizing Fe–Nx clusters in the mesoporous channels, the interactions between iron and nitrogen can be enhanced, thus, more Fe–Nx active sites with unique structures (square pyramidal planar and square planar structure) may be created.44,46,53 For instance, a variety of mesoporous carbons supported transition metal–N species has been proposed to exhibit superior electrocatalytic activity and stability toward ORR.54–60 As a result, it is crucial to study the fabrication of different structures of Fe–Nx embedded in the mesoporous carbons by using task-specific ionic liquids with strong coupling abilities.
In the present work, a series of Fe–Nx doped mesoporous carbons (denoted as FeAMC-T and FeEMIC-T) is synthesized via simultaneous heat-treatment of nitrogen-containing ionic liquids and iron precursors using various carbonization temperatures in mesoporous silicas (i.e., SBA-15). Moreover, ORR performance of FeAMC-T and FeEMIC-T catalysts is evaluated by using linear scan voltammetry (LSV) and rotation disk electrode (RDE) systems. Consequently, FeAMC-1273 possesses not only the best ORR activity but also methanol-tolerant durability in acid media among all prepared catalysts. Furthermore, X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) are employed to explore the molecular-scale structures that may influence the ORR electrocatalytic activity in the FeAMC-T and FeEMIC-T catalysts.
Experimental
Electrocatalysts preparation
Firstly, a mesoporous silica template, namely SBA-15, was prepared by using the route in the earlier report.61 Secondly, two different amino-functionalized ionic liquids were synthesized in this study. In terms of synthesis of (1-(3-aminopropyl)-3-methylimidazolium bromide) ([AM][Br]) ionic liquids, ca. 8.8 g of 3-bromopropylamine hydrobromide and 3.2 mL of 1-methylimidazole were introduced to 100 mL ethanol and then refluxed at 353 K with nitrogen flow for 24 h. The resulting solution was additionally treated by re-crystallization process from ethanol and their corresponding solid product was attained after drying in a vacuum atmosphere at 333 K for 12 h. In terms of preparation of 1-ethyl-3-methylimidazolium dicyanamide ([EMI][DCA]) ionic liquids, the mixture of 1-ethyl-3-methyl-imidazolium-chloride and sodium dicyanamide was stirred at 363 K under dark and nitrogen environment for 5 h. After continually stirring at 323 K for 12 h, dichloromethane (DCM, Acros, 99%) was added and kept at 273 K for 12 h, followed by removal of organic solvents at 373 K in a vacuum oven for 12 h. Thirdly, the direct transformation of silica template into Fe–N supported mesoporous carbons (using the [AM][Br] and [EMI][DCA] ionic liquids and their corresponding notations are FeAMC-T and FeEMIC-T, respectively, where T represents the carbonized temperatures) were accomplished via a simple method as below. In a typical run, fixed amounts of FeCl3 (Acros) and 0.5 g of dehydrated SBA-15 were mixed in absolute ethanol and then dried under vacuum condition. Afterward, the resultant solids were carbonized by using different temperatures (i.e., 873, 1073 and 1273 K) in a nitrogen flow. At last, the resulting solids were washed with 1 wt% HF aqueous solution for 24 h to remove the silica template, then dried at 333 K to obtain the FeAMC-T and FeEMIC-T (T = 873, 1073 and 1273 K).
Electrocatalysts characterizations
Nitrogen sorption isotherms were recorded by using a Micromeritics ASAP 2020 station at 77 K. Elemental analyses (EA) for nitrogen contents were performed by using an Elementar Vario EL III analyzer. Powder X-ray diffraction (XRD) was collected on an X-ray diffractometer (PANalytical, X'Pert PRO) using CuKα radiation (λ = 0.1541 nm). The diffractograms were recorded in the 2θ range from 15 to 90° at a scanning speed of 10° min−1. High-resolution transmission electron microscopy (TEM) was performed with JEOL JEM-2100F field-emission source transmission electron microscope operating at 200 kV. The averaged iron nanoparticle size distribution was estimated by measuring the size of at least one hundred randomly chosen particles in the magnified TEM images. The surface compositions of catalysts were characterized by X-ray photoelectron spectroscopy (XPS) on a spectrometer (Kratos Axis Ultra DLD) with an Al-Kα radiation. The X-ray absorption spectroscopies (XAS) of Fe K-edge (7112 eV) were measured at the Wiggler beamlines 17C of the National Synchrotron Radiation Research Center (NSRRC) in Taiwan. The Fe K-edge of the catalysts was collected at room temperature in transmission mode with a double-crystal Si(111) monochromator to reduce the higher order harmonics in the beam. The XAS data were analyzed by using the package of ATHENA and ARTEMIS software.
Electrocatalytic measurements
The tests of electrocatalytic performance of the catalysts were carried out on a CHI727D electrochemical workstation (CH Instruments, USA) with a rotating disk electrode (RDE) system (Pine Instrument, AFMSRCE). All electrochemical measurements were conducted at room temperature on a three-electrode cell. An Ag/AgCl with saturated KCl and a platinum wire were used as reference and counter electrodes, respectively. To prepare the working electrode, ca. 5 mg of catalyst was dispersed in 2.5 mL deionized water and the mixture was sonicated for 30 min to obtain the homogeneous catalyst ink. An aliquot (ca. 20 μL) of the ink was then deposited on the surface of glassy carbon electrode (GC, 5 mm) and dried at 333 K for 60 min. Furthermore, to confirm the better linkage of the ink on the GC, 20 μL of 1 wt% Nafion® ionomer solution was deposited onto the layer of catalyst ink. Linear sweep voltammetry (LSV) was conducted by using RDE technique, which was performed in oxygen saturated 0.5 M H2SO4 electrolyte at the scan rate of 5 mV s−1. To evaluate the number of electron involved during the ORR, different electrode rotation speed (100–2000 rpm) and potentials (−0.1 to 0.1 V vs. Ag/Ag/Cl) were investigated.
Results and discussion
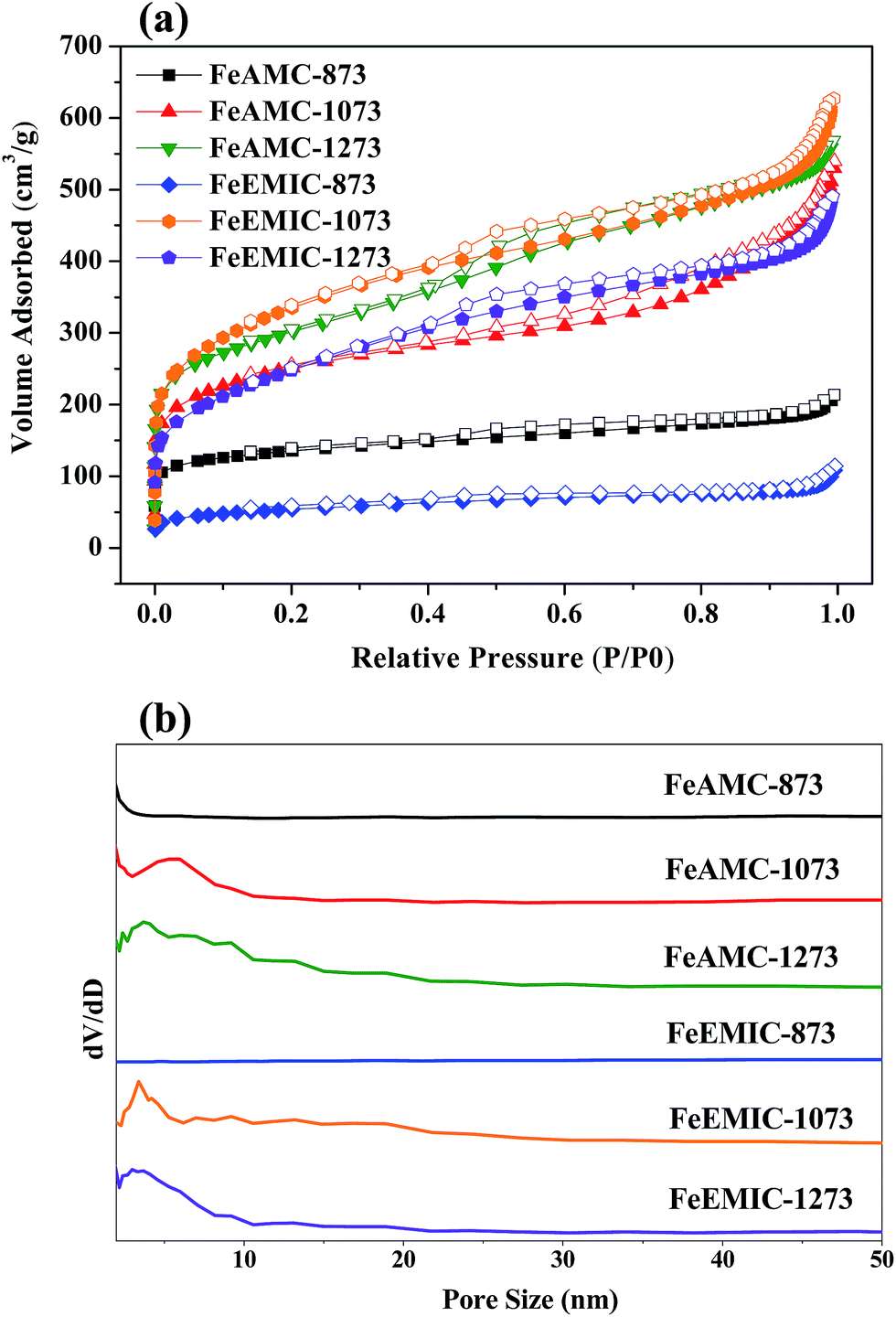
In order to understand physicochemical properties of the FeAMC-T and FeEMIC-T catalysts, a variety of different spectroscopic and analytical techniques was used. The N2 adsorption/desorption curves (see Fig. 1a) and their structural parameters (see Table 1) obtained from FeAMC-873 and FeEMIC-873 catalysts show that the much lower sorption curves and the broad and featureless pore size distribution (see Fig. 1b), indicate the presence of an amorphous carbon composite with a low BET surface area (S) of only ca. 431 and 180 m2 g−1, respectively. However, upon increasing the carbonized temperatures, FeAMC-T (T = 1073 and 1273) and FeEMIC-T (T = 1073 and 1273) catalysts show the type IV characteristic with a sudden increase in the adsorption branch at higher relative pressures, suggesting the formation of mesoporous configuration. Also, the pore size distributions of FeAMC-T (T = 1073 and 1273) and FeEMIC-T (T = 1073 and 1273) catalysts are centered in the mesoporous range of 2.4–5.0 nm. Among them, the FeAMC-1273 catalysts possess a high BET surface area of 1002 m2 g−1 and large pore volume of 0.88 cm3 g−1. However, the nitrogen content of FeAMC-1273 catalysts is found to be ca. 2.8 wt% (Table 1) by elemental analysis (EA), which much less than those of FeAMC-T (T ≤ 1073) catalysts.
 |
| | Fig. 1 (a) N2 adsorption–desorption isotherms and (b) their corresponding pore size distributions of the FeAMC-T and FeEMIC-T catalysts. | |
Table 1 Kinetic parameters and textural properties of FeAMC-T and FeEMIC-T catalysts
| Sample |
S (m2 g−1) |
V (cm3 g−1) |
d (nm) |
N (wt%) |
Eonset (V) |
jmass (mA mgcatalyst−1) |
n |
| SBA-15 |
800 |
1.37 |
10.1 |
— |
— |
— |
— |
| FeAMC-873 |
431 |
0.33 |
— |
17.5 |
0.50 |
∼0 |
4.0 |
| FeAMC-1073 |
822 |
0.83 |
5.0 |
6.5 |
0.67 |
2.85 |
4.0 |
| FeAMC-1273 |
1002 |
0.88 |
4.1 |
2.8 |
0.69 |
5.11 |
— |
| FeEMIC-873 |
180 |
0.18 |
— |
29.4 |
— |
∼0 |
3.7 |
| FeEMIC-1073 |
1131 |
0.96 |
4.0 |
14.8 |
0.64 |
1.67 |
4.0 |
| FeEMIC-1273 |
869 |
0.76 |
2.4 |
2.1 |
0.61 |
1.65 |
4.0 |
As shown in Fig. 2a, the powder XRD patterns show that all the catalysts exhibit a very broad peak at 26° which can be ascribed to the amorphous carbon produced by carbonizing ionic liquids at high temperatures. By comparison, no noticeable diffraction features of iron species are observed for FeAMC-T and FeEMIC-T catalysts, which are probably due to their tiny particle sizes, low contents or amorphous structures of iron compounds. Accordingly, the physical properties of iron in the environment of FeAMC-T and FeEMIC-T catalysts are scarcely unveiled by XRD.
 |
| | Fig. 2 (a) Powder XRD patterns of FeAMC-T and FeEMIC-T catalysts, TEM images of (b) FeAMC-1273, (c) FeEMIC-1273, and (d) HRTEM images of FeAMC-1273. | |
The morphology of iron nanoparticles in the FeAMC-1273 and FeEMIC-1273 catalysts was additionally examined by TEM. As displayed in Fig. 2b, the FeAMC-1273 catalysts with iron nanoparticles of ca. 5–10 nm are uniformly dispersed on the surface of mesoporous carbons. On the contrary, the iron nanoparticles on the FeEMIC-1273 catalysts are to some extent aggregated and not uniform (see Fig. 2c). Furthermore, the iron nanoparticles embedded in mesoporous carbons are examined by the high-resolution TEM, as shown in Fig. 2d. The lattice d-spacing of FeAMC-1273 catalysts is ca. 0.23 nm that is a little bit greater compared to the typical value of 0.203 nm (the (110) crystal plane of metallic iron). This is possibly attributed to the introduction of nitrogen species into the lattices of iron, resulting in the increase of the lattice constant.62 According to combined results obtained from EA, TEM, and N2 sorption isotherms, it is indicative that the fabrication of FeAMC-T catalysts by carbonizing [AM][Br] ionic liquids and iron precursors in the mesoporous silica templates leads to a well-dispersed distribution and small size of iron on the surface of mesoporous carbons with moderate N content, which can enhance greater accessibility of oxygen to these active species.
The surface chemical structure of these N species in various FeAMC-T and FeEMIC-T catalysts were further studied by XPS, as shown in Fig. S1 (ESI†). The high-resolution XPS spectra at N 1s region of FeAMC-T and FeEMIC-T catalysts display three different peaks,62–64 corresponding to pyridinic-N, pyridinic-N–Fe and quaternary-N. Among these species, pyridinic-N and pyridinic-N–Fe play the two important roles in terms of the enhanced ORR performance. As a result, further deconvolution is employed to assess the relative N components on the surfaces of FeAMC-T and FeEMIC-T catalysts, which are summarized in Table 2. Among these catalysts, the total amounts of pyridinic-N and pyridinic-N–Fe observed for FeAMC-1273 catalysts are increased evidently to the maximum (i.e., 87.2%), which may illustrate the best ORR performance of FeAMC-1273, as mentioned below.
Table 2 Percentage content of different N species in total N amount for the FeAMC-T and FeEMIC-T catalysts
| Sample |
Pyridinic-N (%) |
Pyridinic-N–Fe (%) |
Quaternary-N (%) |
| FeAMC-873 |
56.5 (398.4 eV) |
14.3 (399.3 eV) |
29.2 (400.4 eV) |
| FeAMC-1073 |
40.6 (398.2 eV) |
38.6 (399.3 eV) |
20.8 (400.7 eV) |
| FeAMC-1273 |
34.9 (398.3 eV) |
52.3 (399.2 eV) |
12.8 (400.8 eV) |
| FeEMIC-873 |
45.8 (398.4 eV) |
13.7 (399.4 eV) |
40.5 (400.5 eV) |
| FeEMIC-1073 |
26.6 (398.3 eV) |
18.9 (399.3 eV) |
54.5 (400.7 eV) |
| FeEMIC-1273 |
16.2 (398.7 eV) |
22.3 (399.6 eV) |
61.5 (400.7 eV) |
To additionally investigate the nature (oxidation state and geometry) of iron active sites in the FeAMC-T and FeEMIC-T catalysts, the X-ray absorption spectroscopy (XAS) measurements have been also conducted and their corresponding normalized K-edge X-ray absorption near-edge structural (XANES) spectra of FeAMC-T and FeEMIC-T catalysts were demonstrated in Fig. 3a. In addition, standard compounds such as Fe metal, Fe2O3, and FeCl3 are also shown in Fig. 3b. Obviously, the profiles of the FeAMC-T and FeEMIC-T catalysts in the XANES regions are not the same as that of the FeCl3 compounds, implying that the iron precursors (FeCl3) used in the synthesis process have been converted to diverse structures of iron upon reacting with [AM][Br] and [EMI][DCA] during carbonized procedures. Except for the FeEMIC-1273 catalysts (more similar to Fe metal), it can be found that the curves and their corresponding intensities of FeAMC-T and FeEMIC-T catalysts in the XANES spectra are in the middle of metallic (Fe) and oxidized iron (Fe2O3), indicating that the existence of oxidative form of iron in these catalysts. As can be seen in the inset of Fig. 3a, the pre-edge feature at ca. 7114 eV can be attributed to a quadrupole-allowed 1s to 3d transition, which is representative of square-pyramidal Fe(III) complexes.65 It can be observed that the pre-edge peak of FeAMC-1273 is more evident when compared to those of FeAMC-T and FeEMIC-T, indicating the coordination around iron may be square pyramidal structure45 in the FeAMC-1273. This is in good accordance with the extended X-ray absorption fine structural (EXAFS) results discussed below. In terms of EXAFS, Fourier transformations (FT) of the k3-weighted EXAFS functions for FeAMC-T and FeEMIC-T catalysts and standard compounds are shown in Fig. 3c and d, respectively. It should be noted that the radial distance distributions of FeAMC-T and FeEMIC-T catalysts are totally different from those of standard compounds, revealing that diverse local structures may be formed. However, the radial distance distribution of FeEMIC-1273 is more similar to that of Fe metal. In general, the major peaks for FeAMC-T and FeEMIC-T catalysts in the FT spectra (except for FeEMIC-1273) at about 1.4 Å can be assigned to the first shells of Fe–N bonding. No apparent peaks can be observed in the region of higher coordination shells. This indicates the presence of Fe–Nx species in the FeAMC-T and FeEMIC-T catalysts (except for FeEMIC-1273), which are in good agreement with the XPS results. As such, FeAMC-T and FeEMIC-T catalysts (except for FeEMIC-1273) have a majority of Fe–N bonding via carbonizing amino-functionalized ionic liquids ([AM][Br] and [EMI][DCA]) that can intensely chelate with iron precursors and then remain after high-temperature carbonization process. In terms of FeEMIC-1273, however, a strong feature ascribed to Fe–Fe bonding can be found, suggesting the formation of metallic iron at a carbonized temperature of 1273 K. Furthermore, curve fitting process is performed by using FEFF codes from Fe–N and Fe–Fe paths to separate EXAFS contributions from different coordination shells and compute the coordination numbers (CN) nearby the iron atoms. As a result, EXAFS parameters (i.e., CN, bond distance (R), Debye–Waller factor (σ2), and inner potential shift (ΔE)) are summarized in Table 3. Among FeAMC-T and FeEMIC-T catalysts, FeAMC-1273 catalysts possess the average R and CN of 1.98 Å and 4.8, respectively, suggesting the possible formation of square pyramidal planar around iron45 which is in good accordance with the XANES results. By contrast, the FeEMIC-1273 catalysts mainly have a Fe–Fe bond distance of 2.5 Å with a CN of 5.1, implying that the iron in the FeEMIC-1273 is more likely existed as metallic form. Among all catalysts, the FeAMC-1273 catalysts with greater number of N ligands, which can make up the lost electrons transferring to oxygen during ORR, should be accountable for the best ORR performance (see below).
 |
| | Fig. 3 Fe K-edge XANES spectra of (a) various FeAMC-T and FeEMIC-T catalysts and (b) reference compounds, Fourier transform of k3-weighted χ(k) Fe K-edge EXAFS spectrum for (c) various FeAMC-T and FeEMIC-T catalysts and (d) Fe reference compounds. | |
Table 3 EXAFS structural parameters derived from fitting results at Fe K-edge for the FeAMC-T and FeEMIC-T catalysts
| Sample |
Shell |
CN |
R (Å) |
σ2 × 10−3 (Å2) |
ΔE (eV) |
| FeAMC-873 |
Fe–N |
3.7 |
2.01 |
8.2 |
−3.6 |
| FeAMC-1073 |
Fe–N |
3.4 |
2.00 |
5.2 |
−6.3 |
| FeAMC-1273 |
Fe–N |
4.8 |
1.98 |
8.8 |
−9.9 |
| FeEMIC-873 |
Fe–N |
3.0 |
1.98 |
6.3 |
−8.9 |
| FeEMIC-1073 |
Fe–N |
2.7 |
2.01 |
5.0 |
−4.0 |
| FeEMIC-1273 |
Fe–N |
0.8 |
1.93 |
2.6 |
−13.3 |
| Fe–Fe |
5.1 |
2.50 |
9.6 |
−12.3 |
The oxygen reduction performance of FeAMC-T and FeEMIC-T catalysts has been studied by linear scanning voltammetry (LSV) tests which were carried out in O2 saturated 0.5 M H2SO4 solution at room temperature. The LSV curves of FeAMC-T and FeEMIC-T catalysts and a commercial JM-20% Pt/C (Alfa Aesar-Johnson Matthey) are displayed in Fig. 4a and their resultant onset potentials (Eonset) and kinetic mass current densities (jmass at 0.5 V vs. Ag/AgCl) are also listed in Table 1. It can be seen that almost no ORR activity is observed for the FeEMIC-873 catalysts. However, the FeAMC-873 catalysts have an Eonset of 0.50 V vs. Ag/AgCl. When the carbonized temperature is increased to 1073 K, the Eonset and jmass of FeAMC-1073 and FeEMIC-1073 can be increased to 0.67 V (jmass = 2.85 mA mgcatalyst−1) and 0.64 V (jmass = 1.67 mA mgcatalyst−1), respectively. Among all the FeAMC-T and FeEMIC-T catalysts, FeAMC-1273 which was carbonized at 1237 K using [AM][Br] as precursors possess the best O2 electrocatalytic reduction performance (Eonset = 0.69 V vs. Ag/AgCl). Additionally, the jmass of FeAMC-1273 (5.11 mA mgcatalyst−1) is superior to those in the earlier studies.66,67 More importantly, the jmass (at 0.5 V vs. Ag/AgCl) of FeAMC-1273 is ca. 65% that of commercial JM-20% Pt/C catalysts. As shown in Fig. 4b (the representative FeAMC-1273 catalysts), further RDE experiments with various rotation rates (100–2000 rpm) were done in an O2-saturated 0.5 M sulfuric acid solution. The corresponding Koutecky–Levich (K–L) plots for FeAMC-T and FeEMIC-T catalysts are displayed in Fig. 4c. As a result, the number of electrons transferred per O2 molecule (n) derived from the slope of the K–L equation68 can be obtained.
| | |
1/j = 1/jK + 1/jD = 1/jK + 1/(Bω1/2)
| (1) |
| | |
B = 0.62nFCODO2/3ν−1/6
| (2) |
where
j,
jK and
jD are the measured current density, kinetic and diffusion-limiting current densities, respectively. The diffusion-limiting current density is determined by the electrode rotation rate (
ω), the
n in the overall reduction reaction, the Faraday constant (
F), the bulk concentration of O
2 (
CO) dissolved in the electrolyte, the diffusion coefficient for O
2 (
DO), and the kinematic viscosity of the electrolyte (
ν). As shown in
Fig. 4c, the
n values may be derived from the slope of the K–L plots. Consequently,
n values of FeAMC-
T and FeEMIC-
T electrocatalysts (
T = 873, 1073 and 1273) were deduced to be 3.7–4.0 (see
Table 1), indicating that O
2 is reduced to H
2O on the FeAMC-
T and FeEMIC-
T electrocatalysts, namely, proceed
via the effective 4-electron transferred route in the ORR process. The methanol crossover through the proton exchange membrane is one of the critical issues for the cathodic catalysts in the DMFCs. As can be seen in
Fig. 4d, a variety of LSV measurements was also performed to study the influence of methanol concentrations (0.5 and 2.0 M) on ORR activities of FeAMC-1273 catalysts in O
2 saturated 0.5 M H
2SO
4 solution at ambient temperature. In addition, the LSV curves of a commercial JM-20% Pt/C are compared as well in
Fig. 4d. Apparently, the current densities of methanol oxidation on JM-20% Pt/C are greater upon increasing the concentrations of methanol. Nonetheless, the peak current regarding the methanol oxidation reaction on FeAMC-1273 in terms of the same methanol concentration is much smaller than that on the commercial JM-20% Pt/C electrocatalyst. This result indicates that the former catalyst has a surpassing tolerance of potential crossover of methanol during ORR in the DMFCs. Moreover, the durability tests of FeAMC-1273 and commercial JM-20% Pt/C catalysts were also performed by evaluating the current loss after prolonged operation (
ca. 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
000 s) at a constant voltage of 0.4 V
vs. Ag/AgCl in a 0.5 M H
2SO
4 electrolyte saturated with O
2 at a rotating rate of 1600 rpm. As shown in Fig. S2 (ESI
†), the commercially available electrocatalyst displays a significant decrease with a relative current loss of nearly 37%. Nevertheless, FeAMC-1273 catalysts maintain a high relative current density of 80% after 10
000 s, suggesting that the FeAMC-1273 electrode has a highly electrocatalytic stability toward ORR in acid media. The surpassing electrocatalytic activity and tolerance toward methanol for ORR on the FeAMC-1273 catalysts are because of the unique structure of Fe–N chelates which were reported in the previous studies.
69–71 From the integrative results of X-ray related spectroscopies (XRD, XPS and XAS), these FeAMC-1273 catalysts were found to has high specific surface area (1002 m
2 g
−1) of mesoporous carbons decorated with the well-dispersed square pyramidal structure of Fe–N
x species in which the coordinated nitrogen is abundant of pyridinic-N and pyridinic-N–Fe active species, are responsible for the superior as well as durable ORR performance compared with the synthesized catalysts.
 |
| | Fig. 4 (a) Polarization curves of FeAMC-T and FeEMIC-T catalysts, (b) linear sweep voltammetric curves of ORR at different rotation rates for FeAMC-1273, (c) Koutecky–Levich plots for FeAMC-T and FeEMIC-T catalysts, and (d) polarization curves for ORR on FeAMC-1273 (solid line) and a commercial JM-20% Pt/C (dash line) with different amounts of methanol in O2 saturated 0.5 M H2SO4 solution. | |
Conclusions
In this study, the synthesis of mesostructured and well-dispersed square pyramidal structure of Fe–N–C catalysts (FeAMC-T and FeEMIC-T) by simultaneously pyrolyzing two nitrogen-rich ionic liquids ([AM][Br] and [EMI][DCA]), iron precursors (FeCl3) and silica template (SBA-15) was reported. The fabricated FeAMC-T and FeEMIC-T catalysts were tested as cathodic electrodes and their corresponding properties were fully examined by different spectroscopic and analytical techniques as well. The data indicate that the FeAMC-1273 catalysts were found to exhibit the surpassing electrocatalytic properties, such as the best ORR performance (Eonset = 0.69 V vs. Ag/AgCl), four-electron transferred mechanism and methanol-tolerance stability. Results obtained from XRD, N2 sorption isotherms and TEM images show that well-distributed iron particles (ca. 5–10 nm) are embedded in the FeAMC-1273 catalysts with high surface area (1002 m2 g−1) of mesoporous carbons which can enhance reactant/product diffusion. In addition, XPS indicates that the total amounts of pyridinic-N (34.9%) and pyridinic-N–Fe (52.3%) active sites are highest in the FeAMC-1273 catalysts. Moreover, coordination geometry around iron was most likely as the square pyramidal planar structure in the FeAMC-1273 catalysts, as verified by XAS, may be the active sites for the outstanding ORR activity with methanol-resistant durability, which is also comparable to that of a typically available JM-20% Pt/C catalyst (20 wt% Pt on Vulcan XC-72 activated carbon). As such, the synthesized catalysts could have potential opportunities for practical commercialization in the DMFCs.
Acknowledgements
The authors would like to gratefully acknowledge the Ministry of Science and Technology of Taiwan and Ministry of Economic Affairs (Bureau of Energy) of Taiwan (Grant No. MOST 104-ET-E-006-006-ET) for their financial supports.
Notes and references
- M. Nunes, I. M. Rocha, D. M. Fernandes, A. S. Mestre, C. N. Moura, A. P. Carvalho, M. F. R. Pereira and C. Freire, RSC Adv., 2015, 5, 102919–102931 RSC.
- A. Mehmood, M. A. Scibioh, J. Prabhuram, M. G. An and H. Y. Ha, J. Power Sources, 2015, 297, 224–241 CrossRef CAS.
- R. C. Koffi, C. Coutanceau, E. Garnier, J.-M. Léger and C. Lamy, Electrochim. Acta, 2005, 50, 4117–4127 CrossRef CAS.
- S.-H. Liu and J.-R. Wu, Int. J. Hydrogen Energy, 2012, 37, 16994–17001 CrossRef CAS.
- D. Chen, X. Zhao, S. S. Chen, H. F. Li, X. N. Fu, Q. Z. Wu, S. P. Li, Y. Li, B. L. Su and R. S. Ruoff, Carbon, 2014, 68, 755–762 CrossRef CAS.
- A. Mondal and N. R. Jana, RSC Adv., 2015, 5, 85196–85201 RSC.
- H. J. Liu, M. L. Dou, F. Wang, J. J. Liu, J. Ji and Z. L. Li, RSC Adv., 2015, 5, 66471–66475 RSC.
- S.-H. Liu, F.-S. Zheng and J.-R. Wu, Appl. Catal., B, 2011, 108–109, 81–89 CAS.
- X. Q. Huang, Z. P. Zhao, L. Cao, Y. Chen, E. B. Zhu, Z. Y. Lin, M. F. Li, A. M. Yan, A. Zettl, Y. M. Wang, X. F. Duan and T. Mueller, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- Z. S. Li, B. L. Li, Z. S. Liu, Z. H. Liu and D. H. Li, RSC Adv., 2015, 5, 106245–106251 RSC.
- C. J. Bondue, P. Reinsberg, A. A. Abd-El-Latif and H. Baltruschat, Phys. Chem. Chem. Phys., 2015, 17, 25593–25606 RSC.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- G. Sievers, S. Mueller, A. Quade, F. Steffen, S. Jakubith, A. Kruth and V. Brueser, J. Power Sources, 2014, 268, 255–260 CrossRef CAS.
- Y. Feng, F. Ye, H. Liu and J. Yang, Sci. Rep., 2015, 5, 16219 CrossRef CAS PubMed.
- X. Wang, M. Vara, M. Luo, H. W. Huang, A. Ruditskiy, J. Park, S. X. Bao, J. Y. Liu, J. Howe, M. F. Chi, Z. X. Xie and Y. N. Xia, J. Am. Chem. Soc., 2015, 137, 15036–15042 CrossRef CAS PubMed.
- O. O. Fashedemi and K. I. Ozoemena, RSC Adv., 2015, 5, 22869–22878 RSC.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- R. Liu, C. von Malotki, L. Arnold, N. Koshino, H. Higashimura, M. Baumgarten and K. Muellen, J. Am. Chem. Soc., 2011, 133, 10372–10375 CrossRef CAS PubMed.
- S.-H. Liu and J.-R. Wu, Microporous Mesoporous Mater., 2013, 170, 150–154 CrossRef CAS.
- S. Ganesan, N. Leonard and S. C. Barton, Phys. Chem. Chem. Phys., 2014, 16, 4576–4585 RSC.
- P. Song, Y. Wang, J. Pan, W. L. Xu and L. Zhuang, J. Power Sources, 2015, 300, 279–284 CrossRef CAS.
- E. Farjami, M. A. Rottmayer and L. J. Deiner, J. Mater. Chem. A, 2013, 1, 15501–15508 CAS.
- M. Sun, H. J. Liu, Y. Liu, J. H. Qu and J. H. Li, Nanoscale, 2015, 7, 1250–1269 RSC.
- X. X. Yuan, X. L. Ding, C. Y. Wang and Z. F. Ma, Energy Environ. Sci., 2013, 6, 1105–1124 CAS.
- Y. F. Jiang, L. J. Yang, T. Sun, J. Zhao, Z. Y. Lyu, O. Zhuo, X. Z. Wang, Q. Wu, J. Ma and Z. Hu, ACS Catal., 2015, 5, 6707–6712 CrossRef CAS.
- G. Jo, J. Sanetuntikul and S. Shanmugam, RSC Adv., 2015, 5, 53637–53643 RSC.
- M. Wu, Z. Y. Dou, J. J. Chang and L. L. Cui, RSC Adv., 2016, 6, 22781–22790 RSC.
- S. J. Jiang, Z. Li, H. Y. Wang, Y. Wang, L. N. Meng and S. Q. Song, Nanoscale, 2014, 6, 14262–14269 RSC.
- L. Zhang, K. Lee, C. W. B. Bezerra, J. L. Zhang and J. J. Zhang, Electrochim. Acta, 2009, 54, 6631–6636 CrossRef CAS.
- J. Y. Choi, R. S. Hsu and Z. W. Chen, J. Phys. Chem. C, 2010, 114, 8048–8053 CAS.
- A. G. Kong, Y. Y. Kong, X. F. Zhu, Z. Han and Y. J. Shan, Carbon, 2014, 78, 49–59 CrossRef CAS.
- J. T. Wang, S. Li, G. W. Zhu, W. Zhao, R. X. Chen and M. Pan, J. Power Sources, 2013, 240, 381–389 CrossRef CAS.
- F. J. Pérez-Alonso, M. Abdel Salam, T. Herranz, J. L. Gómez de la Fuente, S. A. Al-Thabaiti, S. N. Basahel, M. A. Pena, J. L. G. Fierro and S. Rojas, J. Power Sources, 2013, 240, 494–502 CrossRef.
- L. Q. Jiang, M. Li, L. Lin, Y. F. Li, X. Q. He and L. L. Cui, RSC Adv., 2014, 4, 26653–26661 RSC.
- D. Sebastián, A. Serov, K. Artyushkova, P. Atanassov, A. S. Aricò and V. Baglio, J. Power Sources, 2016, 319, 235–246 CrossRef.
- C. W. B. Bezerra, L. Zhang, K. C. Lee, H. Liu, A. L. B. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- J. H. Zagal, S. Griveau, J. Silva, T. Nyokong and F. Bedioui, Coord. Chem. Rev., 2010, 254, 2755–2791 CrossRef CAS.
- I. Kruusenberg, L. Matisen, Q. Shah, A. M. Kannan and K. Tammeveski, Int. J. Hydrogen Energy, 2012, 37, 4406–4412 CrossRef CAS.
- C. Dominguez, F. J. Perez-Alonso, J. L. G de la Fuente, S. A. Al-Thabaiti, S. N. Basahel, A. O. Alyoubi, A. A. Alshehri, M. A. Pena and S. Rojas, J. Power Sources, 2014, 271, 87–96 CrossRef CAS.
- R. Zhang, Y. X. Peng, Z. P. Li, K. Li, J. Ma, Y. Liao, L. R. Zheng, X. Zuo and D. G. Xia, Electrochim. Acta, 2014, 147, 343–351 CrossRef CAS.
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- T. F. Li, Y. X. Peng, K. R. Zhang, L. R. Zheng, D. G. Xia and X. Zuo, J. Power Sources, 2015, 293, 511–578 CrossRef CAS.
- U. I. Kramm, I. Herrmann-Geppert, J. Behrends, K. Lips, S. Fiechter and P. Bogdanoff, J. Am. Chem. Soc., 2016, 138, 635–640 CrossRef CAS PubMed.
- S.-H. Liu and S.-C. Chen, Carbon, 2016, 105, 282–290 CrossRef CAS.
- Y. X. Peng, Z. P. Li, D. G. Xia, L. R. Zheng, Y. Liao, K. Li and X. Zuo, J. Power Sources, 2015, 291, 20–28 CrossRef CAS.
- Z. L. Li, G. L. Li, L. H. Jiang, J. L. Li, G. Q. Sun, C. G. Xia and F. W. Li, Angew. Chem., Int. Ed., 2015, 54, 1494–1498 CrossRef CAS PubMed.
- W. Yang, T. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206–209 CrossRef CAS PubMed.
- Y. Y. She, Z. G. Lu, M. Ni, L. Li and M. K. H. Leung, ACS Appl. Mater. Interfaces, 2015, 7, 7214–7221 CAS.
- O. Tzialla, G. Kakosimos, C. Athanasekou, E. Galata, G. E. Romanos, G. Pilatos, L. F. Zubeir, M. C. Kroon, B. Iliev, T. J. S. Schubert and K. G. Beltsios, Microporous Mesoporous Mater., 2016, 223, 163–175 CrossRef CAS.
- G. G. Eshetu, M. Armand, H. Ohno, B. Scrosati and S. Passerini, Energy Environ. Sci., 2016, 9, 49–61 CAS.
- Z. Li, J. Liu, Z. Huang, Y. Yang, C. Xia and F. Li, ACS Catal., 2013, 3, 839–845 CrossRef CAS.
- P. Su, H. Xiao, J. Zhao, Y. Yao, Z. Shao, C. Li and Q. Yang, Chem. Sci., 2013, 4, 2941–2946 RSC.
- R. Murugavel, M. G. Walawalkar, M. Dan, H. W. Roesky and C. N. R. Rao, Acc. Chem. Res., 2004, 37, 763–774 CrossRef CAS PubMed.
- J. B. Xu, T. S. Zhao and L. Zeng, Int. J. Hydrogen Energy, 2012, 37, 15976–15982 CrossRef CAS.
- H. W. Liang, W. Wei, Z. S. Wu, X. L. Feng and K. Mullen, J. Am. Chem. Soc., 2013, 135, 16002–16005 CrossRef CAS PubMed.
- J. Y. Cheon, T. Kim, Y. Choi, H. Y. Jeong, M. G. Kim, Y. J. Sa, J. Kim, Z. H. Lee, T. H. Yang, K. J. Kwon, O. Terasaki, G. G. Park, R. R. Adzic and S. H. Joo, Sci. Rep., 2013, 3, 2715 Search PubMed.
- A. G. Kong, B. Dong, X. F. Zhu, Y. Y. Kong, J. L. Zhang and Y. K. Shan, Chem.–Eur. J., 2013, 19, 16170–16175 CrossRef CAS PubMed.
- A. G. Kong, X. F. Zhu, Z. Han, Y. Y. Yu, Y. B. Zhang, B. Dong and Y. K. Shan, ACS Catal., 2014, 4, 1793–1800 CrossRef CAS.
- D. S. Yang, D. Bhattacharjya, M. Y. Song, F. Razmjooei, J. Ko, Q. H. Yang and J. S. Yu, ChemCatChem, 2015, 7, 2882–2890 CrossRef CAS.
- D. S. Yang, M. Y. Song, K. P. Singh and J. S. Yu, Chem. Commun., 2015, 51, 2450–2453 RSC.
- S.-H. Liu, S.-C. Chen and W.-H. Sie, Int. J. Hydrogen Energy, 2011, 36, 15060–15067 CrossRef CAS.
- K. Parvez, S. B. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. L. Feng and K. Mullen, ACS Nano, 2012, 6, 9541–9550 CrossRef CAS PubMed.
- G. Wu, C. M. Johnston, N. H. Mack, K. Artyushkova, M. Ferrandon, M. Nelson, J. S. Lezama-Pacheco, S. D. Conradson, K. L. More, D. Myers and P. Zelenay, J. Mater. Chem., 2011, 21, 11392–11405 RSC.
- M. Q. Wang, W. H. Yang, H. H. Wang, C. Chen, Z. Y. Zhou and S. G. Sun, ACS Catal., 2014, 4, 3928–3936 CrossRef CAS.
- C. Cartier, M. Momenteau, E. Dartyge, A. Fontaine, G. Tourillon, A. Michalowicz and M. Verdaguer, J. Chem. Soc., Dalton Trans., 1992, 609–618 RSC.
- C. H. Choi, S. Y. Lee, S. H. Park and S. I. Woo, Appl. Catal., B, 2011, 103, 362–368 CrossRef CAS.
- S.-H. Liu, J.-R. Wu, C.-J. Pan and B.-J. Hwang, J. Power Sources, 2014, 250, 279–285 CrossRef CAS.
- Y. Qin, J. Li, J. Yuan, Y. Kong, Y. X. Tao, F. R. Lin and S. Li, J. Power Sources, 2014, 272, 696–702 CrossRef CAS.
- S. Baranton, C. Coutanceau, C. Roux, F. Hahn and J.-M. Léger, J. Electroanal. Chem., 2005, 577, 223–234 CrossRef CAS.
- S.-H. Liu and J.-R. Wu, Electrochim. Acta, 2014, 135, 147–153 CrossRef CAS.
- X. J. Huang, Y. N. Luan, P. F. Yao, J. S. Xie, L. Yu, Z. Y. Wu and P. Chen, RSC Adv., 2015, 5, 76599–76606 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: XPS survey spectra of N 1s region for FeAMC-T and FeEMIC-T; current–time (i–t) chronoamperometric response of FeAMC-1273 and commercial JM-20% Pt/C in O2-saturated 0.5 M H2SO4 solution. See DOI: 10.1039/c6ra22549g |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.