
A systematic study on RNA NMR chemical shift calculation based on the automated fragmentation QM/MM approach
Xinsheng Jina,
Tong Zhuab,
John Z. H. Zhangabc and
Xiao He *ab
*ab
aSchool of Chemistry and Molecular Engineering, East China Normal University, Shanghai, 200062, China. E-mail: xiaohe@phy.ecnu.edu.cn
bNYU-ECNU Center for Computational Chemistry, NYU Shanghai, Shanghai, 200062, China
cDepartment of Chemistry, New York University, New York, NY 10003, USA
First published on 7th November 2016
Abstract
1H, 13C and 15N NMR chemical shift calculations on RNAs were performed using the automated fragmentation quantum mechanics/molecular mechanics (AF-QM/MM) approach. Systematic investigation was carried out to examine the influence of density functionals, force fields, ensemble average and explicit solvent molecules on NMR chemical shift calculations. By comparing the performance of a series of density functionals, the results demonstrate that the mPW1PW91 functional is one of the best functionals for predicting RNA 1H and 13C chemical shifts. This study also shows that the performance of the force fields in describing H-bond strength can be validated by AF-QM/MM calculated imino proton chemical shifts. The polarized nucleic acid-specific charge (PNC) model significantly improves the accuracy of imino hydrogen and nitrogen NMR chemical shift prediction as compared to the FF10 force field, which underscores that the electrostatic polarization effect is critical to stabilizing the hydrogen bonds between base pairs in RNAs. Furthermore, the accuracy of the chemical shift of amino proton can be improved by adding explicit water molecules.
1. Introduction
Nuclear magnetic resonance (NMR) spectroscopy is one of the most effective experiments in investigating the structure and dynamics of biological molecules.1,2 Among the parameters measured by NMR, the chemical shifts are the most precise and convenient output which reflects the atomic chemical environment.3 NMR chemical shift plays an important role in structure prediction, validation and refinement for large biomolecules4–16 and molecular crystals.17–19 Recently, the ribonucleic acids have attracted significant interest, such as noncoding ribonucleic acids (ncRNAs) due to its various functions participated in cell activity and gene expression.20,21 In order to study RNA functions, the structure information is usually a prerequisite. As NMR chemical shift is an important parameter for characterizing RNA structure, accurate chemical shift prediction tools need to be developed.Over the past decades, many structure-based approaches have been used in evaluating NMR chemical shifts. Among them there are two main methods: one is the empirical or semi-empirical method based on training a limited number of high-quality NMR structures, such as SHIFTS,22 Altona,23 NUCHEMICS,7,24 PPM,25 and RAMSEY.26 These methods can be applied for nucleic acid NMR chemical shifts prediction. However, they are not well-suited for chemical shift predictions on non-canonical structures, nucleic acid complexes with ligands and protons involved in hydrogen bonding. The other approach for NMR chemical shift prediction is based on quantum mechanical (QM) calculation, which in principle could be applied on any molecular systems. For large biological molecules, full quantum mechanical calculation is still not practical due to its high computational cost. Hence, many economical and efficient methods have been developed in recent years. Cui and Karplus proposed a method for calculating chemical shifts in the QM/MM framework, and concluded that the QM/MM method can provide good description of the environmental effect on chemical shifts.27 Gao et al. also developed a fragment molecular orbital (FMO) method for NMR chemical shift calculations at the Hartree–Fock (HF) level.28,29 Furthermore, Exner and co-workers utilized the adjustable density matrix assembler (ADMA) approach for the QM calculations of NMR chemical shifts on proteins, nucleic acids and protein-nucleic acid complexes.30–32 Tan and Bettens applied the combined fragmentation method (CFM) for protein NMR chemical shift calculations at the QM level.33 In our previous studies, the automated fragmentation quantum mechanics/molecular mechanics (AF-QM/MM) approach was proposed to compute NMR chemical shifts for several proteins with both implicit and explicit solvent models.16,34–38
Apart from protein NMR chemical shift prediction, there are far fewer QM calculations on DNA/RNA chemical shift prediction. Pauwels et al. performed 1H chemical shift calculations on a cross-linked DNA.39 Victoria et al. calculated 1H chemical shifts of DNA, DNA/RNA hybrid and DNA-protein complex using the ADMA method.31 Suardíaz et al. calculated C1′–C5′ RNA ribose chemical shifts using density functional theory.40 Swails et al. predicted 1H, 13C, 15N and 31P chemical shifts of nucleic acids using the automated fragmentation approach.36 However, previous studies have demonstrated that NMR chemical shift calculations on labile protons (imino and amino hydrogens) in RNAs have large deviations from experimental data. The accuracy of labile proton NMR chemical shift prediction for RNAs needs to be further improved.41
In this study, we carried out a systematic study on RNA systems for 1H, 13C and 15N chemical shift predictions using the AF-QM/MM approach. We focused on improving the labile proton chemical shift prediction. The main reason causing the inaccurate prediction of labile proton chemical shifts arises from the deficiency in classical force field and solvation treatment.41,42 Therefore, we employed the polarized nucleic acid-specific charge (PNC) model43,44 in which the electronic polarization effect of the nucleic acid is imbedded in atomic charges that are fitted to electrostatic potential from quantum chemistry calculation. The PNC is shown to improve in the stability of hydrogen bonds between nucleic acids base pairs. We also used explicit solvation model to improve the accuracy of amino hydrogens H22, H42 and H62, whose chemical shifts are highly influenced by solvent.
This paper is organized as follows, the accuracy of AF-QM/MM was first validated for RNA 1H, 13C and 15N chemical shielding calculations as compared to full system QM results. Next, we compared AF-QM/MM calculated 1H, 13C and 15N chemical shifts using implicit solvent model with experimental data. Then, the conformational averaging of chemical shifts based on snapshots from molecular dynamics (MD) simulation were investigated using two sets of force fields. Finally, we discussed the influence of the explicit solvation model on amino hydrogen NMR chemical shift prediction.
2. Computational approaches
2.1. The AF-QM/MM approach
As the basic unit of ribonucleic acid, each nucleotide contains a ribose, a phosphate group and a base (A, C, U, and G). In the AF-QM/MM approach, the entire RNA is divided into nonoverlapping ribonucleotide-centric fragments by cutting the linkage between 3′ carbon atom on the sugar and 3′ oxygen atom on the phosphodiester bond (see Fig. 1). Each ribonucleotide is termed as a core region, other ribonucleotides within a certain range from the core region are considered as the buffer region. Both core region and its buffer region are treated by quantum mechanics, whereas the rest of the atoms is described by Amber FF10 point charge model to account for the electrostatic polarization effect (Fig. 1a). The purpose of the buffer area is to accurately describe the local chemical environment for the core region, such as chemical bonding, base stacking, hydrogen bonding and electrostatic interaction, which can considerably affect the chemical shifts of core region atoms.45,46 Each fragment-centric QM/MM calculation is carried out separately. Only the isotropic shielding constants of the atoms in the core region are extracted from the individual QM/MM calculations. A more detailed illustration of the automated fragmentation scheme is presented in Fig. 1b.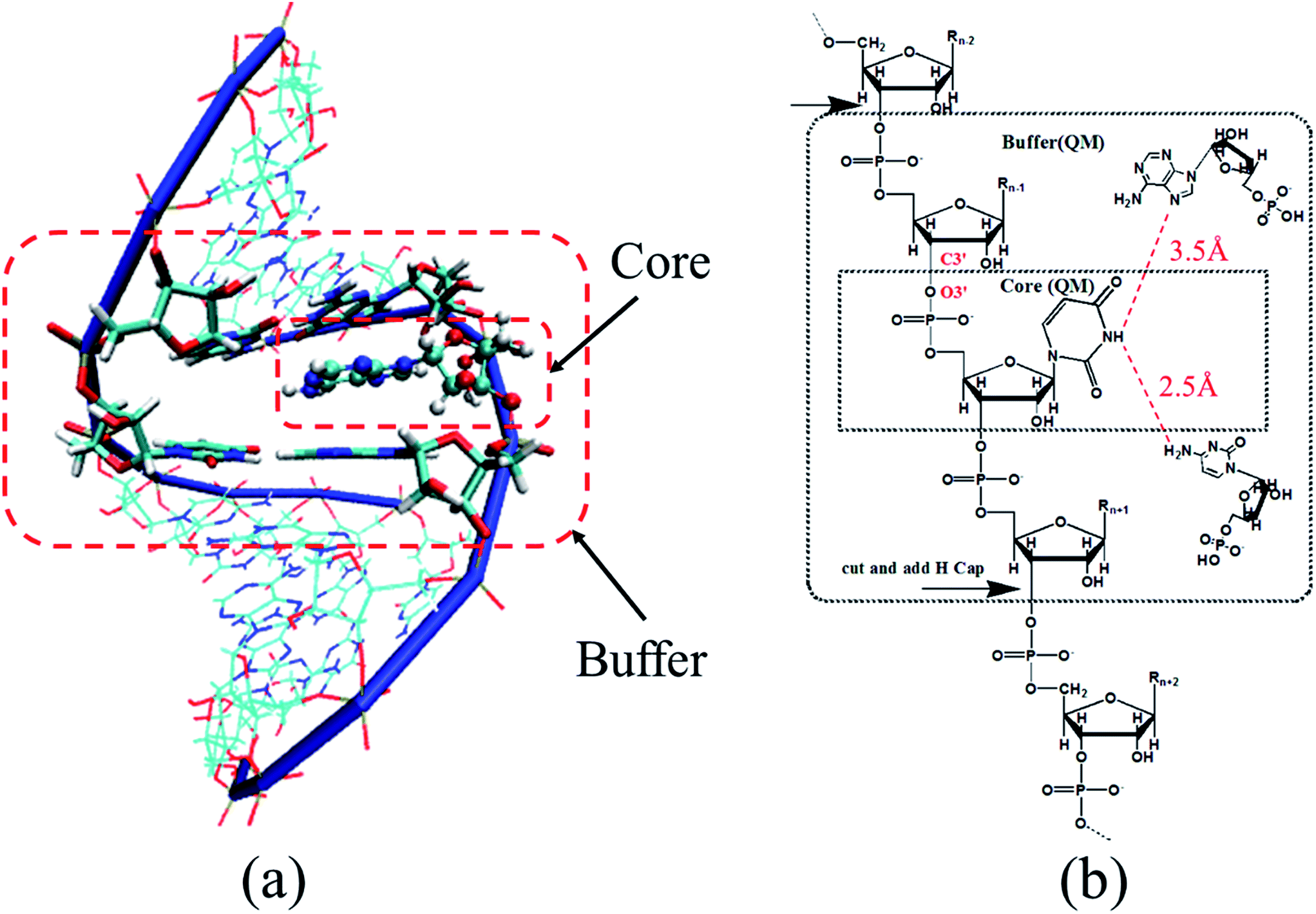 | ||
| Fig. 1 (a) Graphic representation of a nucleotide-centric fragment of RNA duplex (PDB entry: 2K41) in AF-QM/MM calculation. The nucleotide represented by CPK model is the core region, while the neighboring nucleotides and nearby non neighboring nucleotides (which are in close contact with the central nucleotide) represented by thick sticks are in the buffer region. Both the core and buffer regions are treated by QM. The other nucleotides drawn by thin sticks are described by point charges. (b) Definition of the core and buffer regions in AF-QM/MM calculations. | ||
For the nth ribonucleotide, the following three distance-dependent criteria are used to define the buffer region of the nth core region (ribonucleotide): (1) the sequentially connected (n − 1)th and (n + 1)th ribonucleotides (neighboring ribonucleotides); (2) a non-neighboring ribonucleotide outside the core region with an atom less than 3.5 Å away from any atom in the core region (at least one of these two atoms is a non-hydrogen atom); (3) a non-neighboring ribonucleotide outside the core region with a hydrogen atom less than 2.5 Å away from a hydrogen atom in the core region. The non-neighboring ribonucleotides in the buffer region are simply capped by hydrogen atoms to construct the closed-shell fragments (Fig. 1b).
The NMR chemical shift calculation of each fragment was performed by Gaussian 09 (ref. 47) with the GIAO method.48–50 The calculated 1H and 13C chemical shifts are referenced to the isotropic shielding constants of tetramethylsilane (TMS) calculated using each corresponding method (namely, B3LYP, mPW1PW91, OLYP, OPBE, M06 and M062X) with the 6-31G** basis set in gas phase (see Table 1). The reference value for 15N is 265.014 ppm using mPW1PW91/6-31G**, which was obtained from the calculation on ammonia. All calculations were performed in parallel on a Linux cluster with 12-Core Intel Xeon E5645 2.4 GHz processors.
| PDB ID | Method | 1H | 13C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RMSE (ppm) | MUE (ppm) | R2 | Ref. | RMSE (ppm) | MUE (ppm) | R2 | Ref. | ||
| 2K41 | mPW1PW91 | 0.376 | 0.281 | 0.970 | 31.66 | 3.85 | 3.16 | 0.990 | 195.79 |
| B3LYP | 0.378 | 0.284 | 0.970 | 31.76 | 4.41 | 3.51 | 0.988 | 191.82 | |
| M06 | 0.543 | 0.436 | 0.935 | 31.95 | 4.23 | 3.48 | 0.989 | 187.20 | |
| M06-2X | 0.467 | 0.364 | 0.955 | 31.82 | 4.76 | 3.56 | 0.989 | 195.89 | |
| OLYP | 0.410 | 0.308 | 0.970 | 31.72 | 6.56 | 4.71 | 0.982 | 192.17 | |
| OPBE | 0.430 | 0.330 | 0.968 | 31.62 | 6.03 | 4.33 | 0.984 | 196.22 | |
| 2JXS | mPW1PW91 | 0.426 | 0.308 | 0.971 | 4.04 | 3.17 | 0.990 | ||
| B3LYP | 0.436 | 0.317 | 0.970 | 4.75 | 3.62 | 0.987 | |||
| M06 | 0.533 | 0.418 | 0.944 | 4.07 | 3.39 | 0.988 | |||
| M06-2X | 0.448 | 0.358 | 0.959 | 5.03 | 3.77 | 0.988 | |||
| OLYP | 0.479 | 0.348 | 0.967 | 7.34 | 5.18 | 0.979 | |||
| OPBE | 0.487 | 0.363 | 0.967 | 6.75 | 4.80 | 0.982 | |||
2.2. Structure preparation
In this study, we calculated NMR chemical shifts on two double-stranded RNAs (PDB entry: 2K41, with chain A of GUCGUGCUG and chain B of CAGCCGAC; PDB entry: 2JXS, with chain A of GCAGAAGAGCG and chain B of CGCUCUCUGC) and one single-stranded RNA (PDB entry: 1PJY, sequence: GGCCUUCCCACAAGGGAAGGCC). All structures were downloaded from Protein Data Bank. The structures were first optimized using the Amber FF10 force field51,52 of AMBER12 program.53 Since RNA is negatively charged, the geometry optimization of RNA in gas phase will cause longer hydrogen bonds between base pairs. Adding counter ions and explicit solvent (TIP3P water box54) are essential for stabilizing the RNA structure during geometry optimization. The RNA was placed in a truncated octahedral periodic box of TIP3P water molecules. The distance from the surface of the box to the closest atom of the solute was set to 12 Å. Counter ions were added to neutralize the whole system. Two steps of minimizations were employed to optimize the initial structure in explicit water box. In the first step, only the solvent molecules were relaxed, while RNA atoms were constrained to their initial structure. In the second step, the entire system was energy-minimized until convergence was reached.2.3. MD simulation
In MD simulation, the system was heated from 0 to 300 K over 50 ps after geometry optimization. Subsequently, 2 ns MD simulation was performed in the NPT ensemble to further relax the system without any restraints. During MD simulation, the SHAKE algorithm was used to constrain all the bonds involving hydrogen atoms, and the time step was 2 fs. The particle-mesh Ewald method55 was used to treat long-range electrostatic interactions. A 10 Å cutoff for the van der Waals interactions was implemented. The temperature was regulated using Langevin dynamics56 with a collision frequency of 2.0 ps−1. Twenty snapshots from each trajectory were selected every 100 ps for ensemble averaging NMR chemical shift calculations.To further explore the influence of conformational effect simulated by force fields on RNA NMR chemical shift calculations, we compared the results from Amber FF10 and PNC force fields based on ensemble averaging. The PNC charge model provides polarized atomic charges of ribonucleic acids for stabilizing the H-bond between base pairs.44 In MD simulation under PNC, the atomic charges of the Amber FF10 force field were simply replaced by PNC, while the rest of the FF10 force field parameters were retained. In order to have a direct comparison to the FF10 force field, the same MD procedure was applied in PNC simulation.
2.4. Derivation of PNC charges
The PNC charges were fitted to electrostatic potentials by fragment quantum mechanical calculation of RNA in solvent using an iterative approach as described in the molecular fractionation with conjugate caps scheme by incorporating the Poisson–Boltzmann solvation model (MFCC-PB).43 In the MFCC-PB approach, the electron density of each individual nucleotide fragment was calculated at the B3LYP/6-31G* level using the Gaussian09 package. The partial charges of RNA atoms were fitted by a standard two-stage RESP57 fitting procedure. The electronic polarization effect arising from the rest fragments was considered as point charges in each fragment QM calculation. The PB solver Delphi58 was used to calculate the induced charges on the solute–solvent interface which was defined using a probe radius of 1.4 Å. The grid density was set to 4.0 grids per Å in numerically solving the PB equation with the solvent dielectric constant of 80. The corrected reaction field energy from Delphi using the fitted charges was calculated iteratively until its variation was smaller than a certain criterion. Usually the criterion will be reached within five iterations.2.5. Solvation models
In this study, implicit and explicit solvation models were both utilized and compared for analyzing the impact of different solvent models on the NMR chemical shift calculations, especially for H22, H42 and H62, which may form hydrogen bonds with water molecules. For implicit solvation, we utilized the Delphi program58 by solving the PB equation to perform self-consistent reaction field (SCRF) calculations. The impact of solvent acting on RNA could be represented by PB solved surface charges. These point charges were used as background charges in QM chemical shift calculations.With the explicit solvation model, to accurately predict the positions of explicit solvent molecules around the RNA is a major challenge for NMR chemical shift calculation. Using standard MD simulation is inefficient for sampling the influence of explicit water molecules. In order to converge the flux of solvent and ions throughout the biomolecular interior, a long-time MD simulation is usually inevitable. In this study, the distribution of explicit solvent molecules is corrected by the three-dimensional continuous distribution using a 3D reference interaction site model (3D-RISM).59 The algorithm of the 3D-RISM method is based on statistical mechanics and has been shown to accurately reproduce water distributions at a reduced computational cost.60 In this work, the PLACEVENT program developed by Hirata and co-workers61 was utilized to translate the continuous distributions to explicit water molecules. Previous studies have demonstrated that this program places the water molecules on the highest likelihood location and gives excellent agreement with experimental data.62 Geometry optimization is also performed after explicit water molecules are placed. In the optimization procedure, heavy atoms of RNA were restrained while only water molecules and hydrogen atoms on RNA were relaxed. In order to save computational cost, all the water molecules in the first solvation shell from the core region (within 3.5 Å) were kept in the QM region, while all the rest of the water molecules were represented by point charges. The implicit solvent model was used to represent the bulk solvent effect beyond the explicit solvent boundary (see Fig. 2).
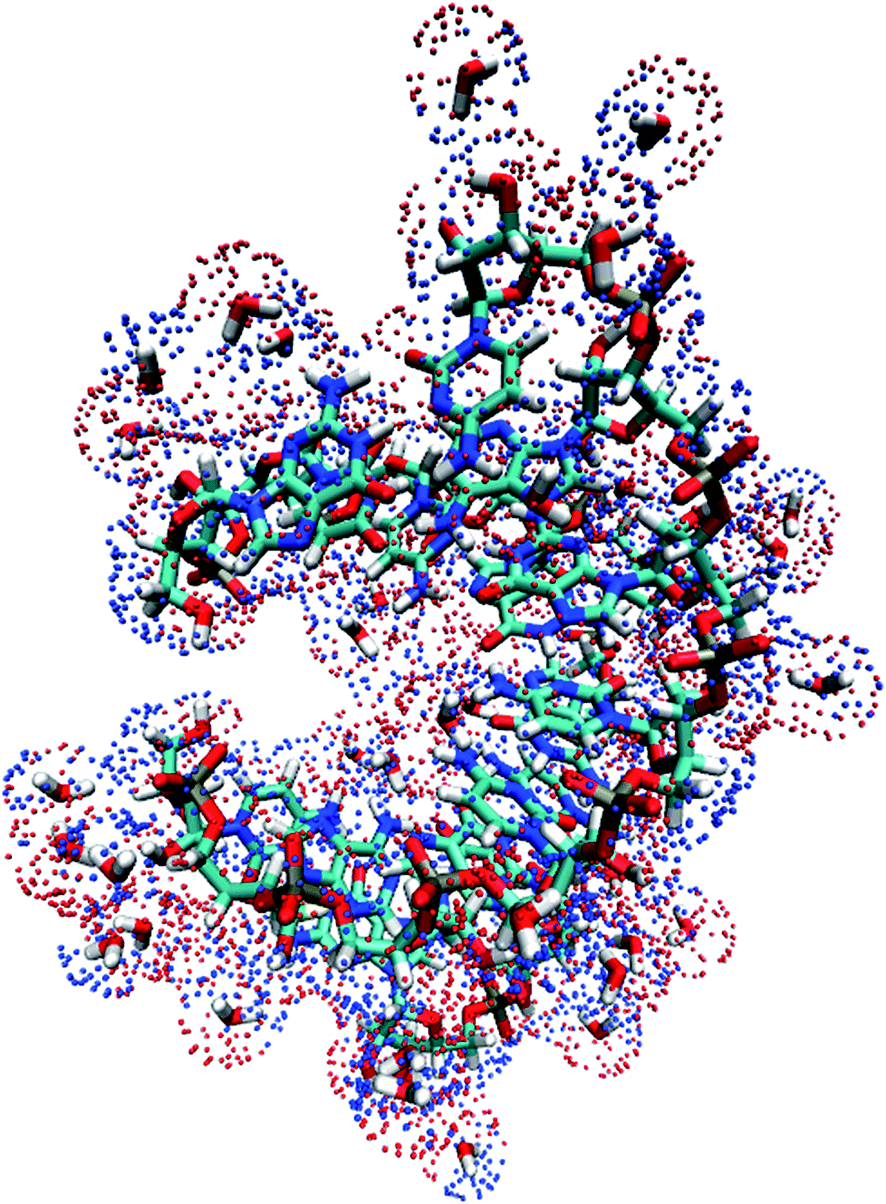 | ||
| Fig. 2 Graphic representation of RNA duplex (PDB entry: 2K41) in explicit solvent environment (within 3.5 Å) and surface charges (implicit solvation beyond the explicit solvation shell) solved by Poisson–Boltzmann equation. Red and blue dots represent the positive and negative surface charges, respectively. | ||
3. Results and discussion
3.1. Benchmark test on a RNA duplex
The AF-QM/MM method is applied to calculate the absolute 1H, 13C and 15N chemical shieldings of the RNA duplex (PDB entry: 2K41, with 17 nucleotides) in gas phase at the mPW1PW91/6-31G** level and compared to the conventional full system results (see Fig. 3). In the full system calculation, the RNA is calculated as an intact molecule without any fragmentation. The RMSEs of 1H, 13C and 15N are 0.14, 0.41 and 1.16 ppm, respectively, with respect to full system results. The performance is similar to that for calculation on proteins.36,37 In addition, the correlation between AF-QM/MM and full system 1H, 13C, 15N NMR chemical shifts are all very close to 1.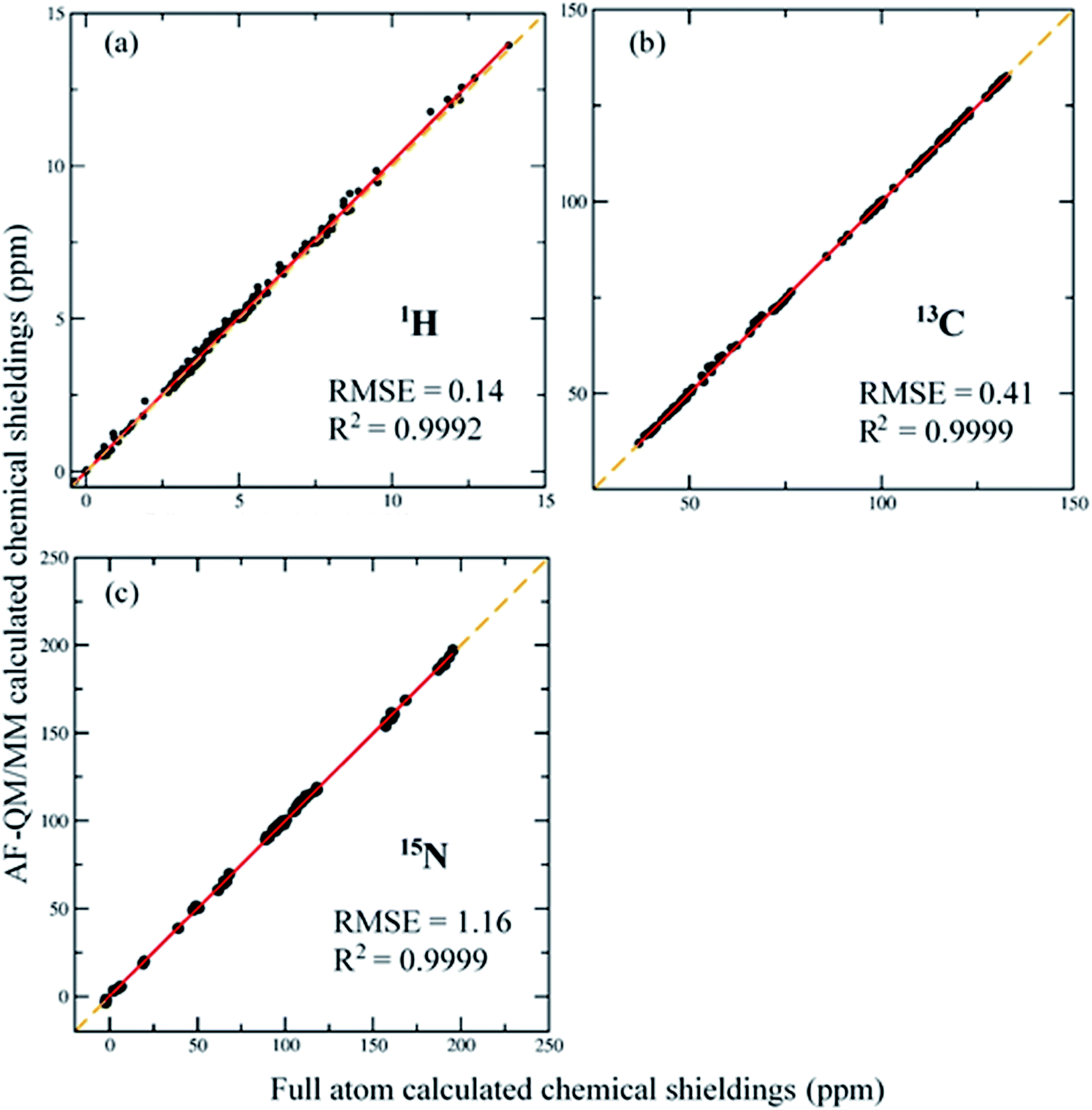 | ||
| Fig. 3 Correlation between conventional full system and AF-QM/MM calculated NMR chemical shifts. 188 proton (a) and 161 carbon (b) and 64 nitrogen (c) atoms' chemical shieldings of RNA 2K41 are compared at the mPW1PW91/6-31G** level. | ||
The comparison clearly demonstrates that the AF-QM/MM calculated chemical shieldings can accurately reproduce the full system results. The benchmark test also shows that current definition of the buffer region is sufficient for accurate calculations of RNA NMR chemical shifts. The average number of atoms in QM fragments of RNA 2K41 is 153 and the largest number of atoms in those fragments is 229. The accuracy of such size of QM sphere was also discussed in previous studies.63,64 The RNA with N ribonucleotides was divided into N ribonucleotide-centric fragments for QM/MM calculations. The size of the QM region is independent with the size of the entire system because each ribonucleotide can only have limited number of ribonucleotides in its vicinity. Therefore, the AF-QM/MM approach is linear-scaling and trivially parallel. The computational wall time of each QM/MM calculation only takes about 1–3 hours on a single node with 12-Core Intel Xeon E5645 2.4 GHz processor.
3.2. Performance of different density functionals
In this section, several density functionals have been tested to investigate their influence on the accuracy of RNA 1H and 13C chemical shifts calculations. Previous studies have shown that density functionals could be one of the factors that may affect the accuracy of chemical shift predictions on organic molecules and proteins.30,35,65 Here we compared the performance of six commonly used density functionals (namely, mPW1PW91, B3LYP, M06, M06-2X, OLYP and OPBE). The test systems are NMR structures of 2K41 and 2JXS. The 6-31G** basis set was employed in all calculations. The calculated results of amino hydrogens H22, H42 and H62 were excluded in comparison with experimental data (see Table 1), because their deviations from experiments would be mostly influenced by the treatment of solvation effect. In this section, the solvation effect was treated with the implicit solvent model. The improvement of NMR chemical shift calculation on these amino hydrogens using the explicit solvation model will be discussed in Section 3.5.As shown in Table 1, the mPW1PW91 functional gives the best performance for both 1H and 13C chemical shift calculations as compared to the experiment. For hydrogen atoms, the RMSEs are 0.376 and 0.426 ppm for 2K41 and 2JXS, respectively. For 2K41, the RMSEs of 1H chemical shifts calculated by B3LYP, OLYP and OPBE functionals are slightly worse than mPW1PW91, while Minnesota functionals (M06 and M06-2X) give relatively larger RMSEs. For 13C chemical shifts, the RMSEs calculated by mPW1PW91 are 3.85 and 4.04 ppm, respectively, for 2K41 and 2JXS, which are consistently smaller than other five functionals. The accuracies of different DFT functionals are quite similar for these two test systems. Hence, we conclude that, among these functionals, the mPW1PW91 is more suitable for RNA 1H and 13C chemical shifts calculations. Based on the tests above, we utilized the mPW1PW91 functional with the 6-31G** basis set in all following calculations.
3.3. Comparison with experiment using the implicit solvation model
The 1H, 13C and 15N NMR chemical shifts calculated using AF-QM/MM on two RNA systems (PDB entries: 2K41, 1PJY) are compared with experimental results (see Fig. 4). The AF-QM/MM calculations are based on the optimized structures with the Poisson–Boltzmann solvation model at the mPW1PW91/6-31G** level. As shown in Fig. 4, the calculated 1H, 13C and 15N chemical shifts correlate well with experimental data, except for H22, H42 and H62, which will be treated with the explicit solvation model (see section 3.5). The implicit solvation model is not sufficient for accurate description of the QM effect arising from the hydrogen bond with solvent molecules. When these outliers (H22, H42 and H62) are excluded, the RMSE of calculated 1H chemical shifts was decreased from 0.521 to 0.376 ppm for 2K41. Moreover, for single-stranded RNA 1PJY, the RMSE was also dropped from 0.774 to 0.644 ppm. Fig. 5 shows the 2D plot of RNA Watson–Crick base pairs, where the labile hydrogens are highlighted using the same color as those in Fig. 4. The intramolecular H-bond donors (H1, H3, H21, H41 and H61) have larger chemical shifts than non-labile protons owing to the shielding effect caused by hydrogen bonding.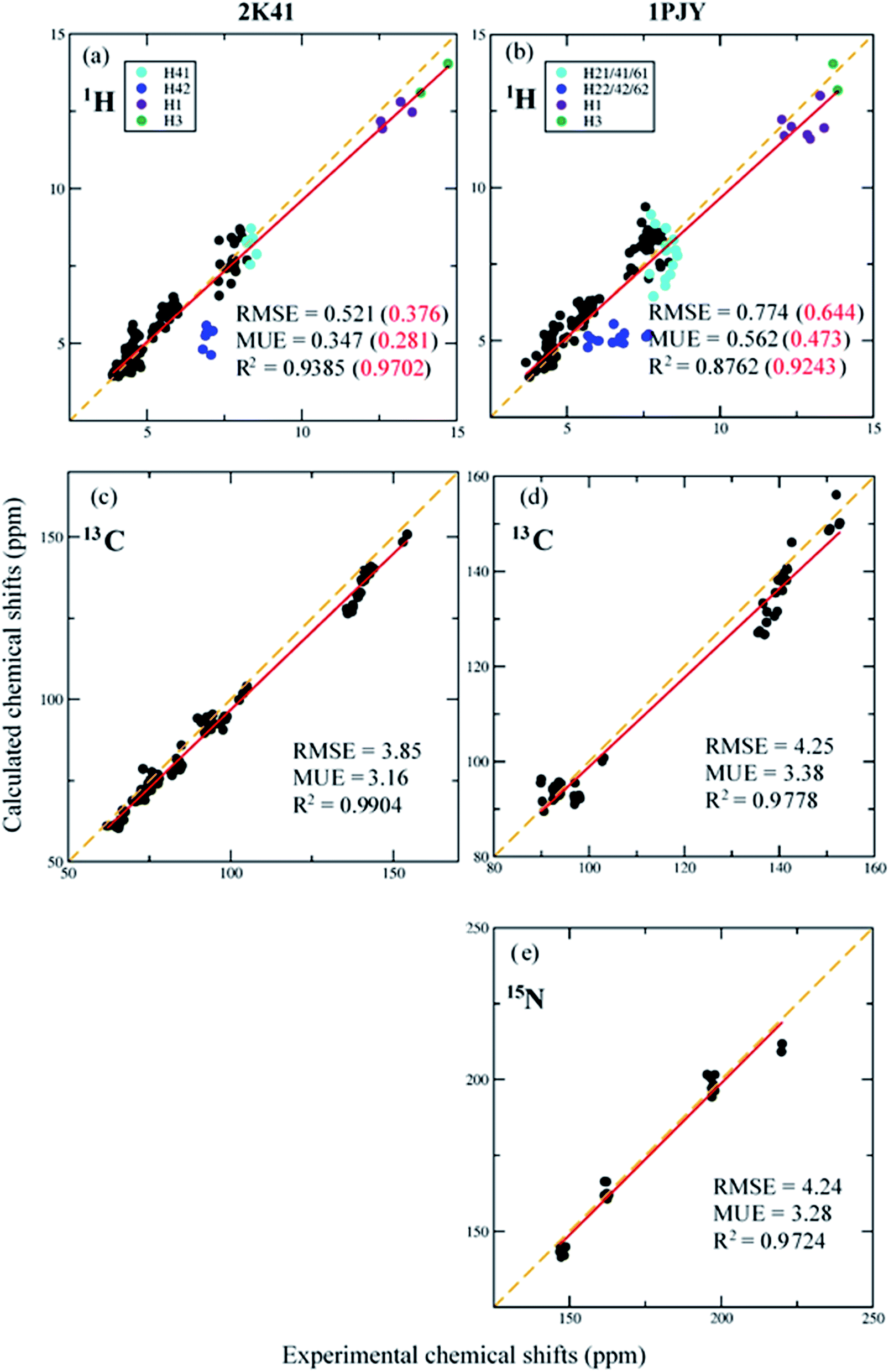 | ||
| Fig. 4 Correlation between experimental and calculated 1H, 13C and 15N chemical shifts of RNA duplex 2K41 (a and c) and single-stranded RNA 1PJY (b, d and e), respectively. In the results of 1H chemical shifts (a and b), the number in parenthesis denotes the result excluding the hydrogen atoms H22, H42 and H62. In the results of 15N chemical shifts for 1PJY, the N7 chemical shifts were neglected. The computational method is mPW1PW91/6-31G**. | ||
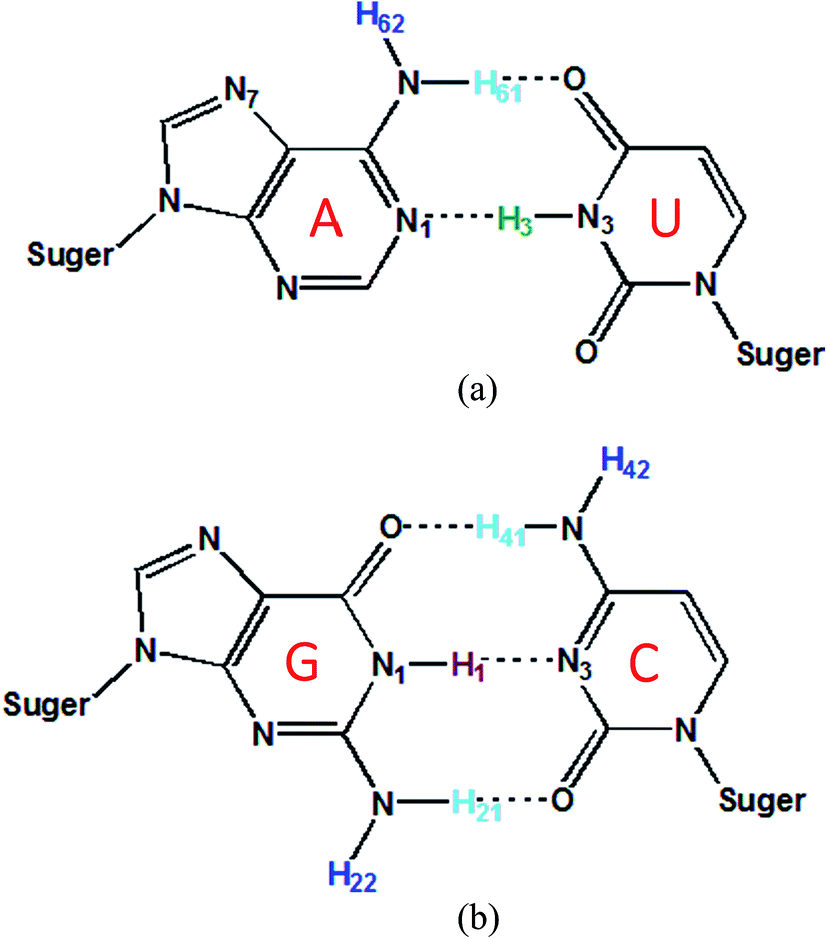 | ||
| Fig. 5 2D plot of Watson–Crick RNA base pairs. The names of hydrogen atoms in the base pair are labeled. Dashed lines denote the hydrogen bonds between base pairs. (a) Adenine-uracil base pair; (b) cytosine-guanine base pair. | ||
For 13C NMR chemical shifts, RMSEs of AF-QM/MM are 3.85 and 4.25 ppm for 2K41 and 1PJY, respectively, with respect to experimental results. As shown in Fig. 4c and d, the AF-QM/MM calculated 13C NMR chemical shifts correlate well with the experiments, where the correlation coefficients (R2) are 0.9904 and 0.9778 for 2K41 and 1PJY, respectively. Fig. 4e shows that the calculated N1 and N3 (defined in Fig. 5) chemical shifts of 1PJY are also in good agreement with the experiment (RMSE = 4.24 ppm and R2 = 0.9724). Therefore, the AF-QM/MM approach with implicit solvation is capable of accurate prediction of 1H (excluding H22, H42 and H62), 13C and 15N NMR chemical shifts for RNAs.
3.4. Ensemble averaging
Although the AF-QM/MM approach captures most static effects on NMR chemical shifts, it is also necessary to take RNA dynamics into consideration in chemical shift calculation, since any measured chemical shift represents the time- and ensemble-average over fluctuations in the RNA structure. As mentioned above, the hydrogen bond between base pairs usually becomes longer and weaker during MD simulations, due to lack of electronic polarization effect in traditional force fields. In this study, we select 20 snapshots of RNAs 2K41 and 1PJY with an interval of 100 ps from 2 ns MD simulation, after the system was well equilibrated using Amber FF10 force field and PNC charge model, respectively. For each conformation, 1H, 13C and 15N chemical shifts were calculated using the AF-QM/MM method at the mPW1PW91/6-31G** level. In addition, the implicit solvation model was used by solving the PB equation for each configuration.We first investigated the imino hydrogen with intramolecular hydrogen bonds (H1 and H3). For RNA 2K41, the H-bond lengths between Adenine-Uracil (AU), and Cytosine-Guanine (CG) are monitored as a function of simulation time (see Fig. 6). For AU H-bond between ribonucleotides 2 and 16 in 2K41 (see Fig. 6a), the experimental H-bond length (average over 20 models) in the NMR structure is 1.70 Å. However, the mean H-bond length over MD simulation by FF10 force field is 1.99 Å, indicating that the H-bond length was substantially exaggerated during MD simulation. Moreover, one can see from Fig. 6a that the H-bond was unstable and broken frequently. The similar trend was also observed for CG H-bond between ribonucleotides 4 and 14 in 2K41, the mean H-bond length was increased from 1.84 Å in the initial NMR structure to 1.97 Å over MD simulation.
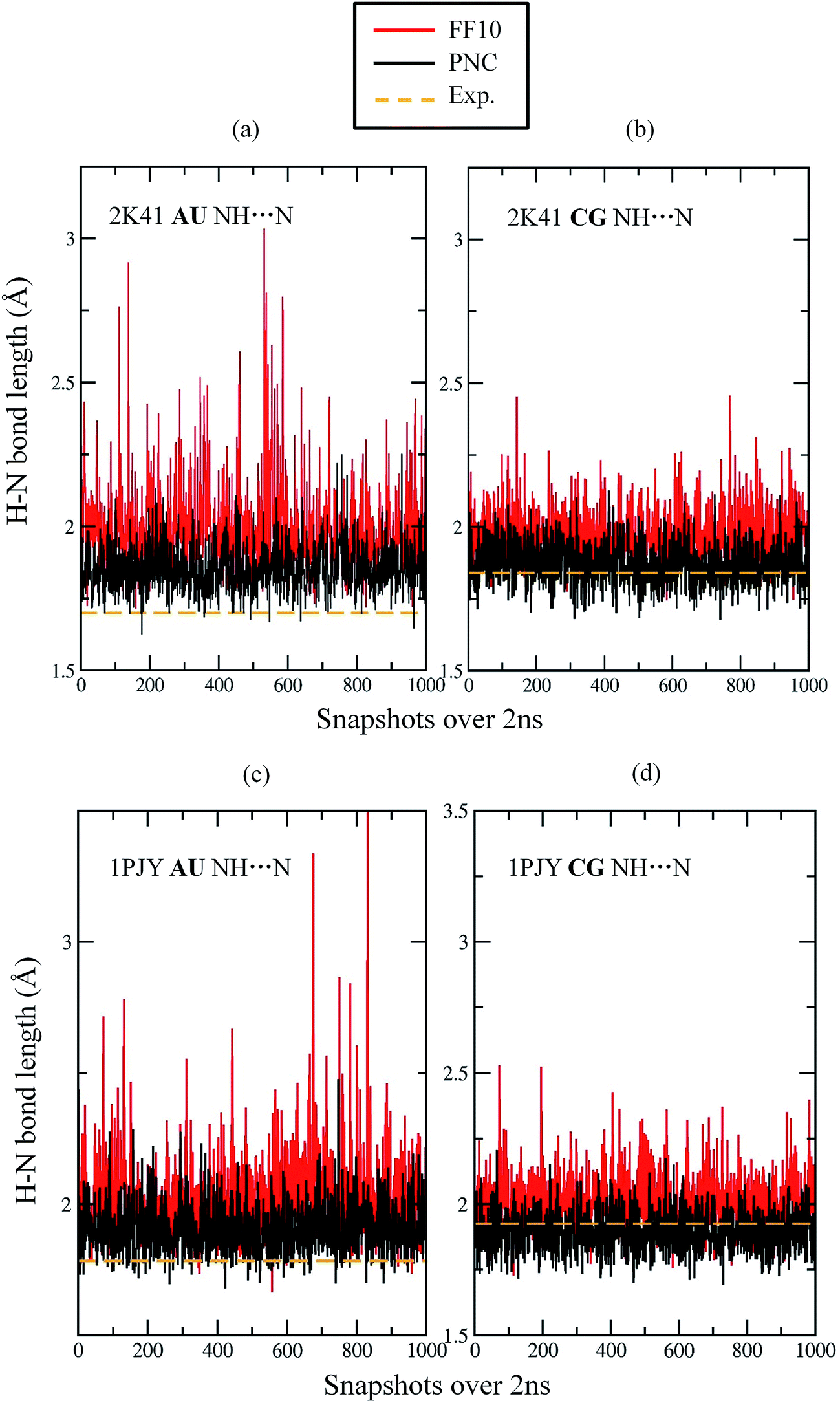 | ||
| Fig. 6 Comparison of H-bond length between base pairs using AMBER FF10 force field (black) and PNC charge model (red), respectively. The dashed line (orange) represents the H-bond length in NMR structures. (a) H-Bond length fluctuation between H3 (in Uracil) of second nucleotide and N1 (in Adenine) of 16th nucleotide of 2K41; (b) H-bond length fluctuation between H1 (in Guanine) of forth nucleotide and N3 (in Cytosine) of 14th nucleotide of 2K41; (c) H-bond length fluctuation between H3 (in Uracil) of sixth nucleotide and N1 (in Adenine) of 17th nucleotide of 1PJY; (d) H-bond length fluctuation between H1 (in Guanine) of first nucleotide and N3 (in Cytosine) of 22nd nucleotide of 1PJY. | ||
On the other hand, the intramolecular H-bond fluctuations in 1PJY are similar to 2K41 under FF10 force field. For instance, the average AU H-bond length between ribonucleotides 6 and 17 is 2.02 Å from MD simulation, as compared to average bond length of 1.78 Å in the NMR structure (see Fig. 6c). In addition, the experimental CG H-bond length between ribonucleotides 1 and 22 is 1.93 Å, while the average length from MD simulation using FF10 is 2.00 Å (Fig. 6d). Therefore, the FF10 force field results in longer and unstable hydrogen bonds between base pairs, which is consistant with Song et al.'s study.44 By taking electronic polarization effect into account, PNC simulation provided shorter and more stable H-bonds as shown in Fig. 6. The H-bond length fluctuation simulated by PNC is much smaller than FF10 force field. The average H-bond lengths given by PNC are closer to the experimental values for all four H-bonds in Fig. 6. The result clearly shows that the electronic polarization is important in stabilizing the H-bond length between RNA base pairs.
The calculated imino proton chemical shifts accurately reflect the deformation of H-bonds across the structure during MD simulation using the nonpolarizable FF10 force field. Because the imino hydrogen chemical shifts are mainly influenced by the H-bond length,46 the increase of the H-bond length will lead to weaker shielding effect that results in a much smaller chemical shift as compared to experiment (see Table 2). As shown in Table 2, the calculated imino hydrogen chemical shifts using PNC are more accurate than those using nonpolarizable FF10 force field. The MUE of H1 chemical shifts in 2K41 decreased from 1.79 (FF10) to 0.57 (PNC) ppm. The improvement of H3 chemical shifts for AU H-bonds is also significant, however, the MUE of 1.37 ppm for H3 atoms using PNC was less accurate than that for H1 in CG base pairs. As shown in Fig. 6, the average AU H-bond length using PNC is above the experimental distance in the NMR structure (Fig. 6a and c), while the CG H-bond length fluctuated around the experimental value in PNC simulation (Fig. 6b and d). Since the CG base pair has three H-bonds, while AU base pair only has two H-bonds, hydrogen bonding of H1–N3 within CG base pair is more stable than H3–N1 in AU base pair. The interaction between the AU base pair under PNC is still not strong enough to accurately maintain the H-bond length during MD simulation, showing the underlying deficiency of the point charge model in describing this particular interaction.
| PDB id | Nucleotide no. | Atom name | FF10a (ppm) | PNCa (ppm) | Exp. (ppm) |
|---|---|---|---|---|---|
| a Deviations from the experiment. | |||||
| 2K41 | 4 | H1 | −1.75 | −0.48 | 12.60 |
| 6 | H1 | −1.59 | −0.26 | 13.19 | |
| 12 | H1 | −1.59 | −0.71 | 13.57 | |
| 15 | H1 | −2.22 | −0.82 | 12.55 | |
| MUE | 1.79 | 0.57 | |||
| 2 | H3 | −3.02 | −1.69 | 14.72 | |
| 8 | H3 | −2.41 | −1.05 | 13.83 | |
| MUE | 2.72 | 1.37 | |||
| 1PJY | 1 | H1 | −2.06 | −1.03 | 12.95 |
| 2 | H1 | −2.16 | −0.90 | 13.40 | |
| 14 | H1 | −1.47 | −0.41 | 12.02 | |
| 15 | H1 | −1.14 | 0.06 | 12.33 | |
| 16 | H1 | −0.65 | −0.12 | 12.10 | |
| 19 | H1 | −1.18 | −0.44 | 12.85 | |
| 20 | H1 | −1.86 | −0.81 | 13.27 | |
| MUE | 1.50 | 0.54 | |||
| 5 | H3 | −2.11 | −1.65 | 13.84 | |
| 6 | H3 | −3.24 | −1.26 | 13.69 | |
| MUE | 2.68 | 1.46 | |||
The comparison between experimental 1H, 13C and 15N NMR chemical shifts and computed ensemble averaging values is shown in Fig. 7. The ensemble averaged 1H chemical shifts from PNC simulation correlate better with experiment than FF10. The RMSEs of 1H chemical shifts by PNC decreased to below 0.5 ppm for both 2K41 and 1PJY. For 13C NMR chemical shifts, the results of PNC were slightly worse than FF10, which may be due to relatively few configurations for ensemble averaging and the limitation of the density functional. For 15N chemical shifts, which only include N1 and N3, the ensemble averaged result of PNC is better than FF10 (the RMSE is reduced from 4.42 to 3.85 ppm), indicating that the hydrogen bonds between base pairs are more accurately described by PNC. By comparing with the results from single structure calculation in Fig. 4, the RMSEs of 13C and 15N NMR chemical shifts are all smaller by taking ensemble averaging using PNC. For 1H chemical shifts, the PNC averaged results give similar performance with single structure calculation when the imino protons are included for 2K41, while ensemble-average results outperform the single structure calculation for 1PJY using both FF10 and PNC force fields.
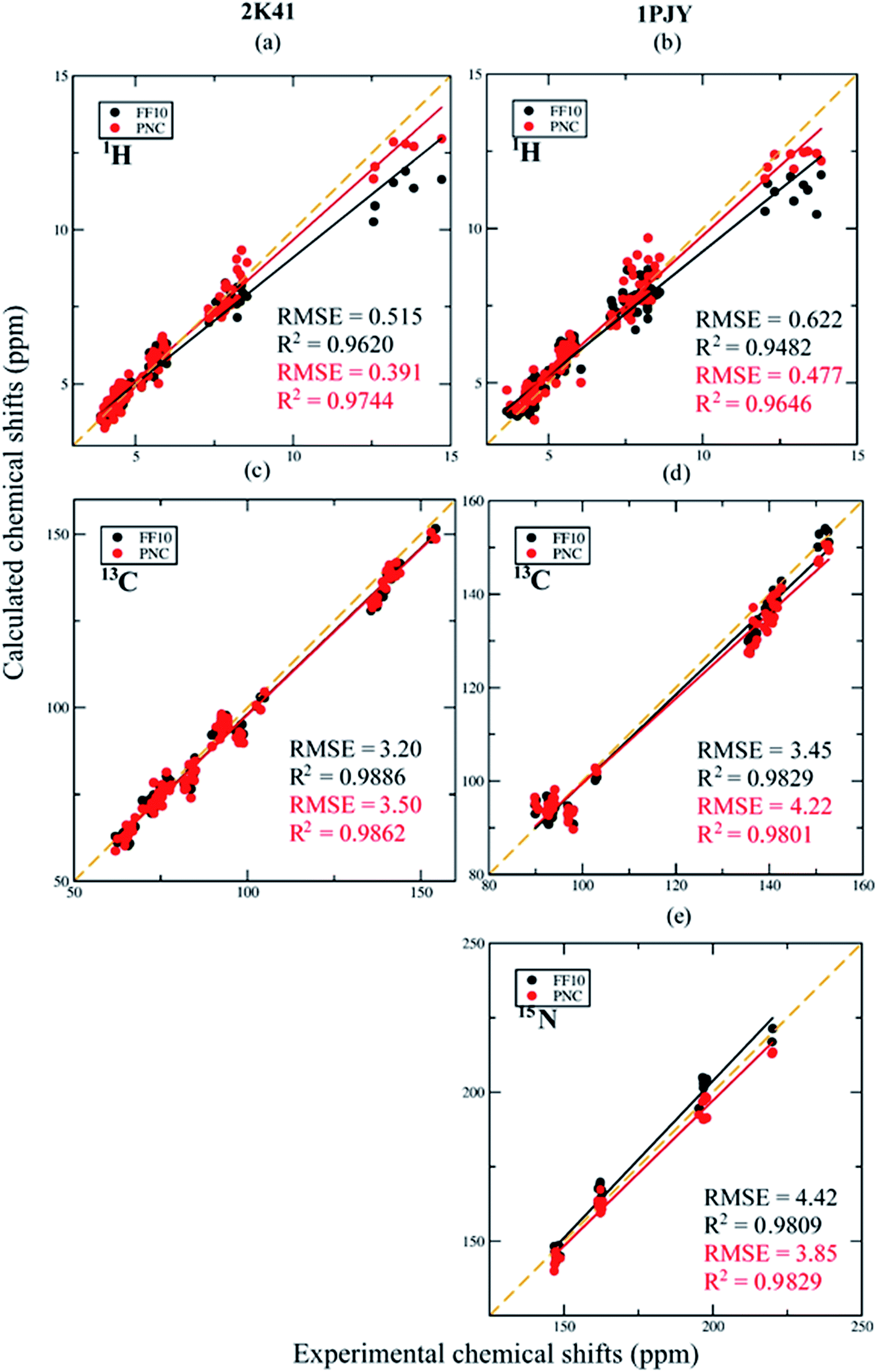 | ||
| Fig. 7 Comparison between ensemble averaging chemical shifts from MD simulations based on Amber FF10 force field and PNC charge model, respectively, for RNA duplex 2K41 (a and c) and single-stranded RNA 1PJY (b, d and e). | ||
3.5. Impact of the explicit solvation model
We also applied explicit solvation model in AF-QM/MM calculations on H22, H42 and H62 chemical shifts for 2K41 and 1PJY, which have largely underestimated chemical shifts as compared to experiments. The deviations mainly arise from the absence of solvent water molecules interacting with those amino protons in RNAs. Fig. 8 shows the relative positions of several key water molecules (predicted by the 3D-RISM method) which have H-bonds with H42 atoms in 2K41. In this study, the inclusion of explicit water molecules gives considerably better agreement for H22, H42 and H62 chemical shifts with experimental results over the implicit model.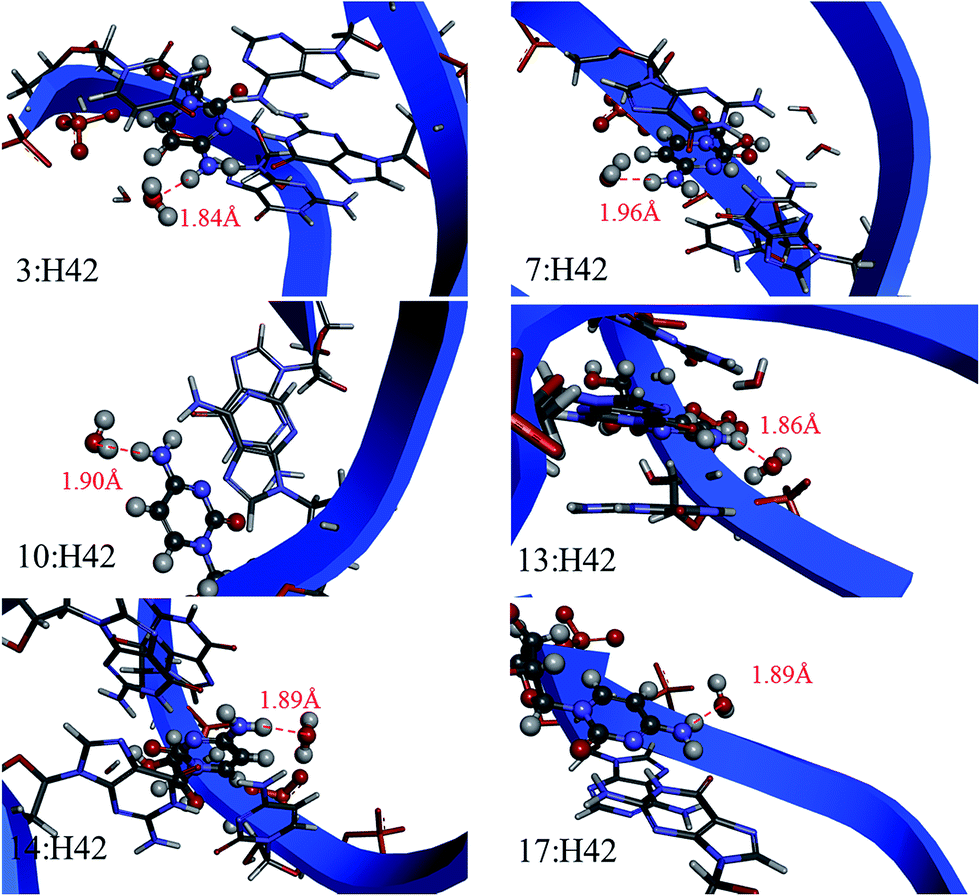 | ||
| Fig. 8 Graphic presentation of explicit water molecules which form hydrogen bonds with H42 in 2K41. The distance is in Å. | ||
Table 3 lists the deviations of H22, H42 and H62 chemical shifts between AF-QM/MM calculation and experimental data when applying implicit and explicit solvent models, respectively. As can be seen from Table 3, the implicit solvation model could not accurately describe the solvent effect of H22, H42 and H62 in NMR chemical shift calculation. When the explicit solvents were included in the fragment QM calculations, the MUE is decreased from 1.55 to 0.80 ppm, as compared to the results using pure implicit solvent model. It clearly shows that hydrogen bonding has a large electronic polarization effect on the H22, H42 and H62 chemical shifts (up to 1–3 ppm). The accuracy of NMR chemical shift prediction for these hydrogen atoms is systematically improved by inclusion of explicit water molecules. However, a few 1H chemical shifts get excessively downfield, which may arise from insufficient sampling of positions of explicit water molecules.
| PDB ID | Nucleotide no. | Atom name | Implicita (ppm) | Explicita (ppm) | Exp. (ppm) |
|---|---|---|---|---|---|
| a Deviations from the experiment. | |||||
| 2K41 | 3 | H42 | −1.34 | 0.37 | 6.92 |
| 7 | H42 | −2.00 | −1.34 | 6.80 | |
| 10 | H42 | −1.72 | −0.82 | 7.12 | |
| 13 | H42 | −1.60 | 0.65 | 6.91 | |
| 14 | H42 | −1.65 | 0.62 | 6.89 | |
| 17 | H42 | −2.41 | −0.09 | 7.07 | |
| 1PJY | 2 | H22 | −2.46 | −0.61 | 7.59 |
| 3 | H42 | −1.94 | −0.45 | 6.85 | |
| 4 | H42 | −1.67 | 0.20 | 6.88 | |
| 7 | H42 | −1.63 | 0.90 | 6.73 | |
| 8 | H42 | −1.74 | −0.66 | 6.71 | |
| 9 | H42 | −0.53 | 1.76 | 5.69 | |
| 14 | H22 | −1.05 | 0.82 | 6.04 | |
| 15 | H22 | −0.73 | 1.03 | 5.78 | |
| 16 | H22 | −0.91 | 1.09 | 5.68 | |
| 17 | H62 | −0.99 | 1.36 | 6.53 | |
| 18 | H62 | −1.56 | 1.30 | 6.49 | |
| 19 | H22 | −1.01 | 1.04 | 6.00 | |
| 20 | H22 | −2.46 | −0.30 | 7.67 | |
| 21 | H42 | −1.64 | 0.58 | 6.88 | |
| MUE | 1.55 | 0.80 | |||
4. Conclusion
In this work, we performed DFT calculations on RNA 1H, 13C and 15N NMR chemical shifts using the AF-QM/MM approach. Our result demonstrates that the mPW1PW91 functional is the best density functional for prediction of RNA 1H and 13C chemical shifts among a selective set of DFT functionals. The NMR chemical shift calculation on RNA with the implicit solvation model scales linearly with the system size. The surface charges solved from the PB equation are applied to each fragment calculation, which avoids separate ab initio solvation calculations on each fragment.The ensemble averaging of 1H, 13C and 15N chemical shifts is also calculated over the snapshots from MD simulation. The imino hydrogen chemical shifts could be used as probes to test the stability of intra-RNA hydrogen bonds. Our study demonstrates that the electronic polarization is critical to stabilizing the H-bonds in RNAs. The PNC model significantly improved the accuracy of imino hydrogen NMR chemical shifts as compared to the FF10 force field. The chemical shifts of nitrogens in H-bond donor or acceptor are also improved by utilizing PNC charge model during MD simulation. The chemical shifts of atoms on intramolecular H-bonds calculated by AF-QM/MM can be utilized as a benchmark test to evaluate the accuracy of molecular force fields in describing the H-bond strength during the MD simulation.
With the implicit solvation model, the accuracy of the AF-QM/MM method on RNA NMR chemical shift prediction is comparable to previous fragment-based QM studies.31,36 As the results indicated the insufficiency of implicit solvation in treating the amino hydrogen atoms, which form H-bonds with solvent molecules, we further investigated the influence of these intermolecular H-bonds on amino proton (H22, H42 and H62) NMR chemical shifts by inclusion of explicit solvent molecules. The result clearly shows a remarkable improvement over that from implicit solvation calculation, without adding much additional computational cost. Moreover, our results demonstrate that the presence of water molecules inside the RNA grooves can be certified using the AF-QM/MM calculated NMR chemical shifts.
In summary, the AF-QM/MM method is able to describe the local chemical environment in RNA. The approach is massively parallel and can be applied to routinely calculate the ab initio 1H, 13C and 15N NMR chemical shifts for RNAs of any size. The PNC and explicit solvation model generally improved the liable proton chemical shifts. In future studies, the AF-QM/MM approach will be applied to more general biological systems such as RNA/DNA-ligand and RNA/DNA-protein binding complexes.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants No. 21303057, 21433004, 21403068 and 21673074), Ministry of Science and Technology of China (Grant no. 2016YFA0501700), Specialized Research Fund for the Doctoral Program of Higher Education (Grant No. 20130076120019), Shanghai Putuo District (Grant 2014-A-02) and NYU Global Seed Grant for Collaborative Research. We also thank the Supercomputer Center of East China Normal University for providing us with computational time.References
- A. H. Kwan, M. Mobli, P. R. Gooley, G. F. King and J. P. Mackay, FEBS J., 2011, 278, 687–703 CrossRef CAS PubMed.
- D. J. Patel, L. Shapiro and D. Hare, Q. Rev. Biophys., 1987, 20, 35–112 CrossRef CAS PubMed.
- D. S. Wishart, B. D. Sykes and F. M. Richards, J. Mol. Biol., 1991, 222, 311–333 CrossRef CAS PubMed.
- A. Cavalli, X. Salvatella, C. M. Dobson and M. Vendruscolo, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9615–9620 CrossRef CAS PubMed.
- Y. Shen and A. Bax, J. Biomol. NMR, 2013, 56, 227–241 CrossRef CAS PubMed.
- H.-b. Le, J. G. Pearson, A. C. de Dios and E. Oldfield, J. Am. Chem. Soc., 1995, 117, 3800–3807 CrossRef CAS.
- J. A. Cromsigt, C. W. Hilbers and S. S. Wijmenga, J. Biomol. NMR, 2001, 21, 11–29 CrossRef CAS PubMed.
- P. Sripakdeevong, M. Cevec, A. T. Chang, M. C. Erat, M. Ziegeler, Q. Zhao, G. E. Fox, X. Gao, S. D. Kennedy, R. Kierzek, E. P. Nikonowicz, H. Schwalbe, R. K. O. Sigel, D. H. Turner and R. Das, Nat. Methods, 2014, 11, 413–416 CrossRef CAS PubMed.
- R. Das, J. Karanicolas and D. Baker, Nat. Methods, 2010, 7, 291–294 CrossRef CAS PubMed.
- J. Ferner, A. Villa, E. Duchardt, E. Widjajakusuma, J. Wöhnert, G. Stock and H. Schwalbe, Nucleic Acids Res., 2008, 36, 1928–1940 CrossRef CAS PubMed.
- J. A. Vila and H. A. Scheraga, Acc. Chem. Res., 2009, 42, 1545–1553 CrossRef CAS PubMed.
- B. S. Tolbert, Y. Miyazaki, S. Barton, B. Kinde, P. Starck, R. Singh, A. Bax, D. A. Case and M. F. Summers, J. Biomol. NMR, 2010, 47, 205–219 CrossRef CAS PubMed.
- J. A. Vila, Y. A. Arnautova, O. A. Martin and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16972–16977 CrossRef CAS PubMed.
- J. A. Vila, Y. A. Arnautova and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1891–1896 CrossRef CAS PubMed.
- A. S. Larsen, L. A. Bratholm, A. S. Christensen, M. Channir and J. H. Jensen, PeerJ, 2015, 3, e1344 Search PubMed.
- T. Zhu, J. Z. Zhang and X. He, Phys. Chem. Chem. Phys., 2014, 16, 18163–18169 RSC.
- D. Stueber, Concepts Magn. Reson., Part A, 2006, 28, 347–368 CrossRef.
- J. D. Hartman and G. J. Beran, J. Chem. Theory Comput., 2014, 10, 4862–4872 CrossRef CAS PubMed.
- D. Solís, M. Ferraro and J. C. Facelli, J. Mol. Struct., 2002, 602, 159–164 CrossRef.
- T. R. Cech and J. A. Steitz, Cell, 2014, 157, 77–94 CrossRef CAS PubMed.
- J. S. Mattick, Hum. Mol. Genet., 2006, 15, R17–R29 CrossRef CAS PubMed.
- X.-P. Xu and D. A. Case, J. Biomol. NMR, 2001, 21, 321–333 CrossRef CAS PubMed.
- C. Altona, D. H. Faber and A. J. Hoekzema, Magn. Reson. Chem., 2000, 38, 95–107 CrossRef CAS.
- S. S. Wijmenga, M. Kruithof and C. W. Hilbers, J. Biomol. NMR, 1997, 10, 337–350 CrossRef CAS PubMed.
- D.-W. Li and R. Brüschweiler, J. Biomol. NMR, 2012, 54, 257–265 CrossRef CAS PubMed.
- A. T. Frank, S.-H. Bae and A. C. Stelzer, J. Phys. Chem. B, 2013, 117, 13497–13506 CrossRef CAS PubMed.
- Q. Cui and M. Karplus, J. Phys. Chem. B, 2000, 104, 3721–3743 CrossRef CAS.
- Q. Gao, S. Yokojima, D. G. Fedorov, K. Kitaura, M. Sakurai and S. Nakamura, J. Chem. Theory Comput., 2010, 6, 1428–1444 CrossRef CAS.
- Q. Gao, S. Yokojima, T. Kohno, T. Ishida, D. G. Fedorov, K. Kitaura, M. Fujihira and S. Nakamura, Chem. Phys. Lett., 2007, 445, 331–339 CrossRef CAS.
- A. Frank, H. M. Möller and T. E. Exner, J. Chem. Theory Comput., 2012, 8, 1480–1492 CrossRef CAS PubMed.
- A. Victora, H. M. Möller and T. E. Exner, Nucleic Acids Res., 2014, 42, e173 CrossRef PubMed.
- T. E. Exner, A. Frank, I. Onila and H. M. Möller, J. Chem. Theory Comput., 2012, 8, 4818–4827 CrossRef CAS PubMed.
- H.-J. Tan and R. P. Bettens, Phys. Chem. Chem. Phys., 2013, 15, 7541–7547 RSC.
- T. Zhu, X. He and J. Z. H. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 7837–7845 RSC.
- T. Zhu, J. Z. H. Zhang and X. He, J. Chem. Theory Comput., 2013, 9, 2104–2114 CrossRef CAS PubMed.
- J. Swails, T. Zhu, X. He and D. A. Case, J. Biomol. NMR, 2015, 1–15 Search PubMed.
- X. He, T. Zhu, X. Wang, J. Liu and J. Z. Zhang, Acc. Chem. Res., 2014, 47, 2748–2757 CrossRef CAS PubMed.
- X. He, B. Wang and K. M. Merz Jr, J. Phys. Chem. B, 2009, 113, 10380–10388 CrossRef CAS PubMed.
- E. Pauwels, D. Claeys, J. C. Martins, M. Waroquier, G. Bifulco, V. Van Speybroeck and A. Madder, RSC Adv., 2013, 3, 3925–3938 RSC.
- R. Suardíaz, A. B. Sahakyan and M. Vendruscolo, J. Chem. Phys., 2013, 139, 034101 CrossRef PubMed.
- T. van Mourik, J. Chem. Phys., 2006, 125, 191101 CrossRef PubMed.
- J. Ho, M. B. Newcomer, C. M. Ragain, J. A. Gascon, E. R. Batista, J. P. Loria and V. S. Batista, J. Chem. Theory Comput., 2014, 10, 5125–5135 CrossRef CAS PubMed.
- C. Ji, Y. Mei and J. Z. Zhang, Biophys. J., 2008, 95, 1080–1088 CrossRef CAS PubMed.
- J. Song, C. Ji and J. Z. H. Zhang, Phys. Chem. Chem. Phys., 2013, 15, 3846 RSC.
- A. B. Sahakyan and M. Vendruscolo, J. Phys. Chem. B, 2013, 117, 1989–1998 CrossRef CAS PubMed.
- A. Grishaev, J. Ying and A. Bax, J. Am. Chem. Soc., 2012, 134, 6956–6959 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. F. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian Inc., Wallingford, CT, 2010.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- A. Pérez, I. Marchán, D. Svozil, J. Sponer, T. E. Cheatham, C. A. Laughton and M. Orozco, Biophys. J., 2007, 92, 3817–3829 CrossRef PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, WIREs Computational Molecular Science, 2013, 3, 198–210 CrossRef CAS.
- W. L. Jorgensen and C. Jenson, J. Comput. Chem., 1998, 19, 1179–1186 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- R. W. Pastor, B. R. Brooks and A. Szabo, Mol. Phys., 1988, 65, 1409–1419 CrossRef.
- C. I. Bayly, P. Cieplak, W. Cornell and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- W. Rocchia, E. Alexov and B. Honig, J. Phys. Chem. B, 2001, 105, 6507–6514 CrossRef CAS.
- T. Imai, A. Kovalenko and F. Hirata, Chem. Phys. Lett., 2004, 395, 1–6 CrossRef CAS.
- T. Imai, R. Hiraoka, A. Kovalenko and F. Hirata, Protein Struct. Funct. Genet., 2007, 66, 804–813 CrossRef CAS PubMed.
- D. J. Sindhikara, N. Yoshida and F. Hirata, J. Comput. Chem., 2012, 33, 1536–1543 CrossRef CAS PubMed.
- T. Imai, K. Oda, A. Kovalenko, F. Hirata and A. Kidera, J. Am. Chem. Soc., 2009, 131, 12430–12440 CrossRef CAS PubMed.
- C. V. Sumowski and C. Ochsenfeld, J. Phys. Chem. A, 2009, 113, 11734–11741 CrossRef CAS PubMed.
- D. Flaig and C. Ochsenfeld, Phys. Chem. Chem. Phys., 2013, 15, 9392–9396 RSC.
- C. Bassarello, P. Cimino, L. Gomez-Paloma, R. Riccio and G. Bifulco, Tetrahedron, 2003, 59, 9555–9562 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |