
Biotransformation of neuro-inflammation inhibitor kellerin using Angelica sinensis (Oliv.) Diels callus†
Abstract
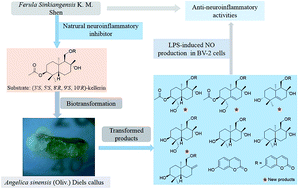
Kellerin, a sesquiterpene coumarin derivative, has been identified as a major constituent of Ferula sinkiangensis K. M. Shen. It has been proved to be a potential natural therapeutic agent for Alzheimer's disease because of its inhibition of inflammatory cytokines nitric oxide (NO), tumor necrosis factor-α (IL-6) (TNF-α), cyclooxygenase-2 (COX-2), interleukin-6 (IL-6) and interleukin-1β (IL-1β) in over-activated BV2 mouse microglial cells. Because of the multi chiral centers and the chemical instability of the sesquiterpene, coumarin, it is rather difficult to obtain this bioactive natural product by synthesis. Thus, biotransformation of kellerin was carried out to afford more novel derivatives using the callus of Angelica sinensis (Oliv.) Diels, which has an abundance of biosynthetic enzymes of phenylpropanoids. As a result, 14 products were obtained and identified, including four new sesquiterpene coumarin derivatives: 14′-hydroxy-(3′S,4′R,5′S,8′R,9′S,10′R)-kellerin (1), 5′,6′-ene-14′-hydroxy-(3′S,4′R,8′R,9′S,10′R)-kellerin (2), 5′,6′-ene-(3′R,8′R,9′S,10′R)-ferukrin (3), and 14′-hydroxy-(3′S,4′R,5′S,8′R,9′S,10′R)-deacetylkellerin (4), together with 10 other known compounds. Their structures were elucidated using comprehensive spectroscopic techniques and their possible biosynthetic pathways were proposed on the basis of the structural analyses. Furthermore, their anti-neuroinflammatory activities were assessed in BV2 cells by monitoring lipopolysaccharide-induced NO production, and the structure–activity relationships were discussed.
 Please wait while we load your content...
Please wait while we load your content...

