DOI:
10.1039/C6RA22480F
(Paper)
RSC Adv., 2016,
6, 96946-96962
Synthetic investigation on chirally pure Mannich derivatives of pseudophenylpropanolamine and their anticancer properties against HepG-2 cells with inhibition of JAK2/STAT3†
Received
8th September 2016
, Accepted 5th October 2016
First published on 7th October 2016
Abstract
A novel series of Mannich derivatives of 3a–g and 3a′–g′ were designed and synthesized from pseudophenylpropanolamine (Ψ-PPA). The stereo chemical aspects of the synthesized compounds were studied and all compounds were well characterized with respect to spectral techniques. All Mannich derivatives, 3a–g and 3a′–g′ were evaluated for their anti-proliferative activity against A549 and HepG-2 cells. Among the tested compounds, 3a showed significant anti-proliferative activity against HepG-2 cells at 25 μM when compared to other compounds. The treatment of 3a exhibited morphological changes, nuclear condensation, colony formatting ability, apoptosis and cell cycle arrest at G2/M phase in HepG-2 cells. Besides, 3a triggered mitochondrial mediated apoptotic pathway as indicated by down regulation of Bcl-2, up-regulation of Bax, and release of cytochrome c and caspases-3. Furthermore, 3a effectively suppressed the cell proliferation and cell growth via JAK2/STAT3 signaling pathway in a time and dose dependent manner. In vivo administration of 3a inhibited tumor growth without significant change in body weight in HepG-2 xenograft mice model. Molecular docking studies revealed that good binding energies of compound 3a against JAK2 (−6.10 kcal mol−1) and Bcl-2 (−6.04 kcal mol−1) receptors. Taken together, 3a possessed potent antitumor activity; it could be a promising lead candidate for the potential treatment of human hepatocellular carcinoma.
Introduction
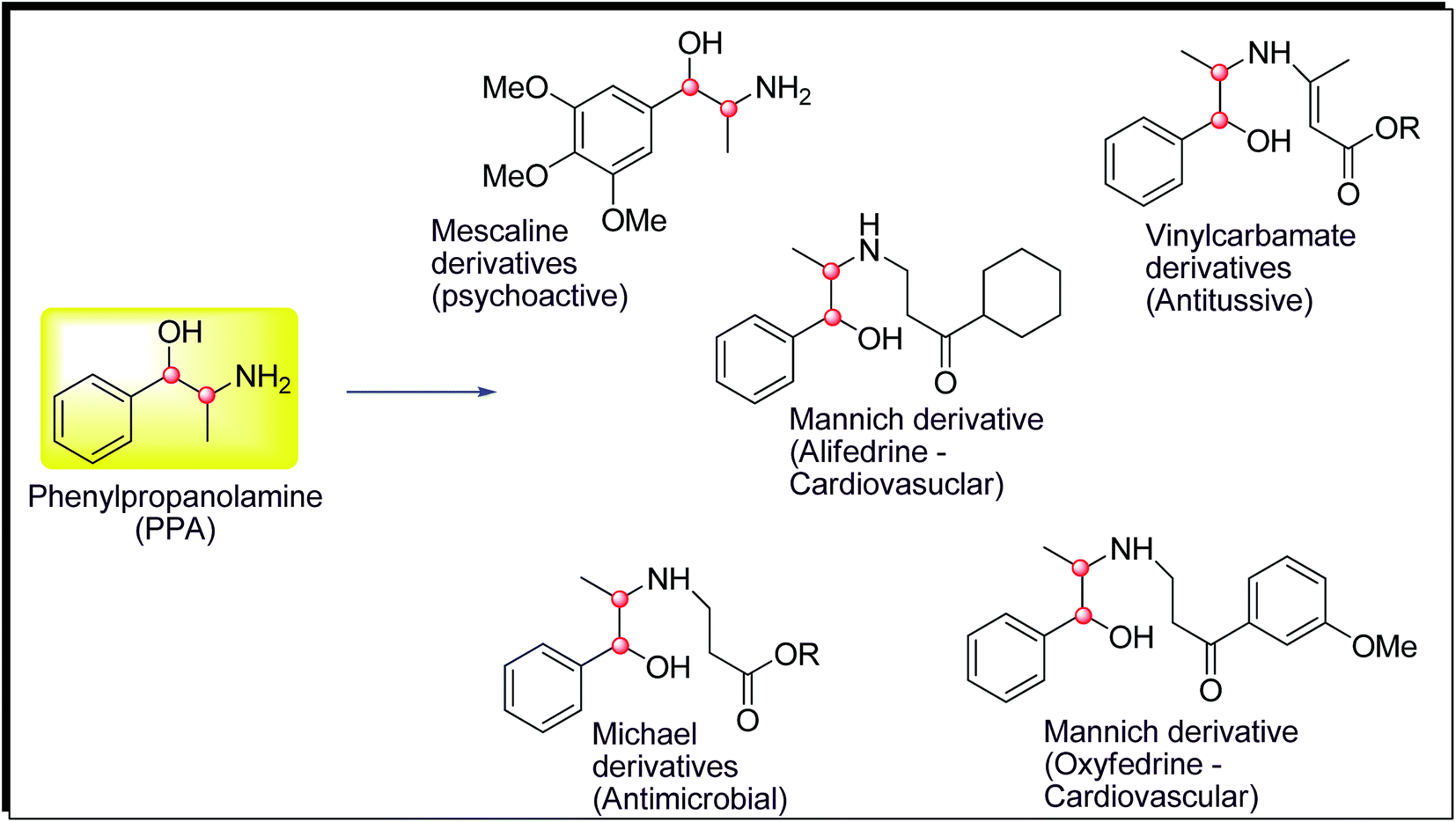
Phenylpropanolamine (PPA or norephedrine) and pseudophenylpropanolamine (Ψ-PPA or norpseudoephedrine) are well known for their psychoactive properties as well as decongestant, stimulant and anorectic effects.1–6 These were used as over the counter (OTC) drugs for decongestant for about 40 years after their invention.7,8 The usage of PPA has been regulated in many countries due to its adverse effects related to cardiovascular, stroke, psychiatric disorders, etc.9–12 Hence, the development of structurally modified PPAs with minimum adverse effect is one of the tasks for researchers. Different types of structurally modified PPA with multiple biological activities such as cardiovascular, antitussive, antimicrobial, antibiotic are known (Fig. 1). Alifedrine and oxyfedrine prepared as Mannich derivative of PPA are known as potential cardiovascular agent and antimicrobial.13–18 Vinyl carbamate derivatives of PPA are known for their antitussive activity.19 Mescaline derivatives of PPA are known as psychotropic agents.20 Based on various important biological activities of derivatives of PPA, herewith we are reporting the synthesis of chirally pure Mannich derivatives of Ψ-PPA as part of our research activity on PPAs.21,22
 |
| | Fig. 1 Various derivatives of phenylpropanolamine. | |
Hepatocellular carcinoma is a liver cancer, also called malignant hepatoma. Hepatocellular carcinoma is the most common form of liver cancer in men and women, particularly in adults.23 Liver cancer is sixth leading cause of cancer deaths worldwide.24 The incidence of hepatocellular carcinoma is highest in Asia and Africa, where the endemic high prevalence of hepatitis B and hepatitis C strongly predisposes to the development of chronic liver disease and subsequent development of hepatocellular carcinoma. Cancer is initiated by uncontrolled cell growth by different environmental conditions such as carcinogens, viruses, chemicals and radiation. Beside, some genetic modifications also occur during cancer such as mutations, malignant growth, invasion and metastasis. Moreover, the treatment of liver cancer uses a combination of different modalities, surgery, chemotherapy, radiotherapy, remission, relapse and metastasis.25 STAT proteins are a family of cytoplasmic transcription factors that are involved in cell proliferation, survival, apoptosis and differentiation.26 More than seven STATs proteins have been discovered from mammalians and particularly STAT3 is most closely linked to cancer development in all human cancers.27 Normally, STAT3 and its upstream JAK signaling were originally identified as the signaling pathway for IFN. It mediates the immune responses of various cytokines as well as many growth factors and hormones; it also participates in inflammation, cell growth and metastasis.28 The IL-6 mediated activation of STAT3 plays a critical role in promoting tumorigenesis.29 STAT3 signaling pathway has recently been shown to confer resistance to chemotherapy induced apoptosis in human tumors, due to its involvement in the proliferation, angiogenesis, immune evasion and anti-apoptosis of cancer cells and its aberrant activation in various tumor cells.30 In the current study, we examined the anticancer properties of synthesized novel Mannich derivatives against A549 and HepG-2 cells using in vivo xenograft mice model.
Results and discussion
Synthesis of chirally pure Mannich derivatives
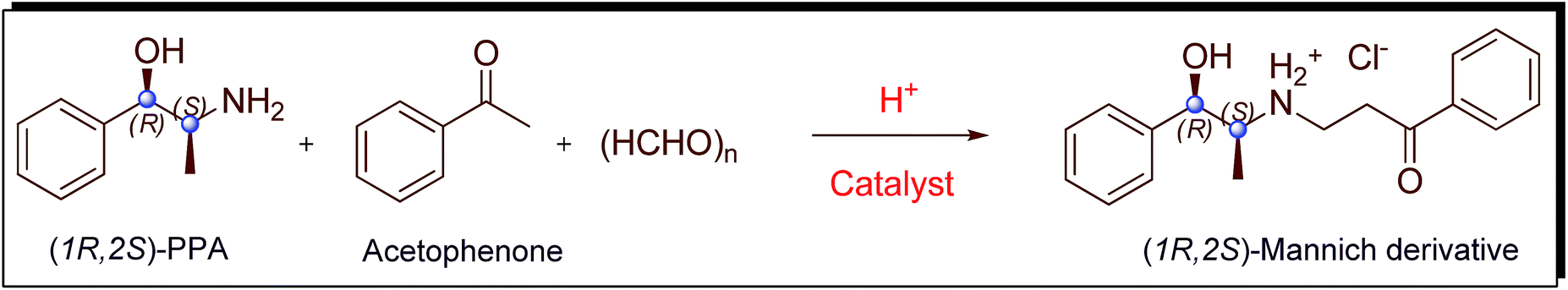
As a continuation of our research work22 on chirally pure Mannich derivatives of PPA, we had reported the synthesis of enantiomerically pure derivatives of PPA using boron trifluoride catalyzed Mannich reaction (Scheme 1). During this study, we had investigated the reason for racemization at benzylic carbon31–33 of (1R,2S)-PPA when using conventional Mannich reaction method16,34–38 which involves heating with hydrochloric acid to prepare (1R,2S)-Mannich derivative from the corresponding PPA (Scheme 1). However, the usage of boron trifluoride had thrown some queries on us, even though the method was capable of producing chirally pure Mannich derivatives of PPA. Toxicology studies of boron trifluoride revealed that, exposure to it resulted in renal toxicity, pneumonities, etc.39 Since the Mannich compounds of PPA are of pharmacological interest, we tried to prepare these without using boron trifluoride as well as with chiral purity. During our study, we developed a method to prepare chirally pure Mannich derivatives of Ψ-PPA using recyclable 10% sulphuric acid immobilized on silica gel (H2SO4·SiO2) instead of toxic boron trifluoride. The Mannich compounds of PPA obtained from these method are found to have chiral purity and in good yields as in the case of boron trifluoride. The outcome details of (1R,2S)-Mannich derivative of PPA prepared from three different methods are given in Scheme 2 and Table 1. During our current study, Mannich reaction catalyzed by the sulphuric acid immobilized on silica gel was extended to prepare chirally pure Mannich derivatives of Ψ-PPA 1 and 1′. As presented in Scheme 3 and Table 2 (1S,2S)- and (1R,2R)-Ψ-PPA (1 and 1′ respectively) were treated with various substituted acetophenones 2a–g, paraformaldehyde in isopropanol medium at 45–50 °C for 6 to 10 h to form the chirally pure pseudo Mannich derivatives 3a–g and 3a′–g′.
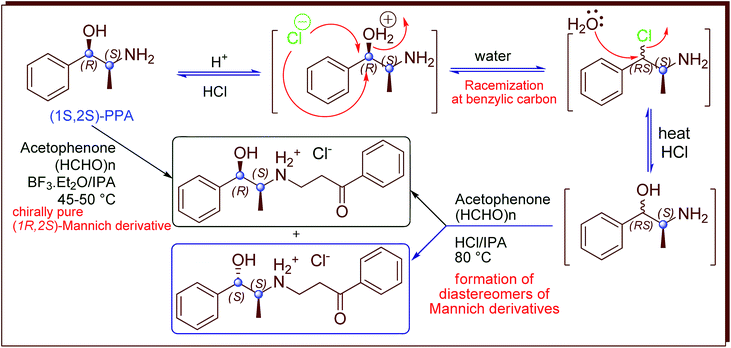 |
| | Scheme 1 Synthesis of Mannich derivatives of (1R,2S)-PPA. | |
 |
| | Scheme 2 Effect of catalysts on formation of chirally pure (1R,2S)-Mannich derivative. | |
Table 1 Effect of catalyst on formation of chirally pure (1R,2S)-Mannich derivative 3a
| Entry |
Catalyst |
Reaction condition |
Purity by ordinary HPLC (area%) |
Enantiomeric purity (ee) by chiral HPLC (area%) |
Yieldb (%) |
| IPA-isopropyl alcohol. Yields as hydrochloride salt of (1R,2S)-Mannich derivative. Diastereomers. Reusable for 6 times. |
| 1 |
HCl34–37 |
75–80 °C in IPA |
Two peaks in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratioc :1 ratioc |
— |
81 |
| 2 |
BF3·Et2O22 (10 mol%) |
45–50 °C in IPA |
99.5 |
99+ |
94 |
| 3 |
10% H2SO4·SiO2d (10 mol%) |
30–35 °C in IPA |
99.7 |
99+ |
95 |
 |
| | Scheme 3 Synthesis of Mannich derivatives 3a–g and 3a′–g′ from pseudoPPA. | |
Table 2 Mannich derivatives of pseudophenylpropanolamine 3a–g and 3a′–g′
|
| Entry |
R |
Product |
Stereo configuration |
Melting point (°C) |
Puritya (%) |
SORb |
eec (%) |
Yieldd (%) |
| Purity by HPLC (area%). Specific optical rotation (c = 1%, methanol). Enantiomeric excess by HPLC. Yield as hydrochloride salt. |
| 1 |
Hydrogen |
3a |
(1S,2S) |
107–110 |
99+ |
+58.5 |
>99.9 |
94 |
| 2 |
Hydrogen |
3a′ |
(1R,2R) |
111–113 |
99+ |
−56.4° |
>99.9 |
90 |
| 3 |
4-Methyl |
3b |
(1S,2S) |
204–206 |
99+ |
+47.1° |
>99.9 |
90 |
| 4 |
4-Methyl |
3b′ |
(1R,2R) |
205–206 |
99+ |
−49.2° |
>99.9 |
90 |
| 5 |
4-Chloro |
3c |
(1S,2S) |
138–141 |
99+ |
+38.8° |
>99.9 |
88 |
| 6 |
4-Chloro |
3c′ |
(1R,2R) |
142–143 |
99+ |
−37.4° |
>99.9 |
87 |
| 7 |
2,4-Dichloro |
3d |
(1S,2S) |
198–200 |
99+ |
+27.8° |
>99.9 |
95 |
| 8 |
2,4-Dichloro |
3d′ |
(1R,2R) |
196–197 |
99+ |
−25.2° |
>99.9 |
89 |
| 9 |
3-Hydroxy |
3e |
(1S,2S) |
172–175 |
99+ |
+57.6° |
>99.9 |
89 |
| 10 |
3-Hydroxy |
3e′ |
(1R,2R) |
174–176 |
99+ |
−58.7° |
>99.9 |
94 |
| 11 |
3-Nitro |
3f |
(1S,2S) |
158–159 |
99+ |
+56.9° |
>99.9 |
92 |
| 12 |
3-Nitro |
3f′ |
(1R,2R) |
164–165 |
99+ |
−57.4° |
>99.9 |
90 |
| 13 |
4-Methoxy |
3g |
(1S,2S) |
155–157 |
99+ |
+40.4° |
>99.9 |
86 |
| 14 |
4-Methoxy |
3g′ |
(1R,2R) |
155–157 |
99+ |
−39.2° |
>99.9 |
87 |
Spectroscopic studies
The structure of all the synthesized compounds (3a–g and 3a′–g′) were confirmed (crystallographic data, 1443803) using spectral analysis such as IR, 1H-NMR, 13C-NMR and ESI-mass spectra (Fig. 2). As a representative, structure of compound 3c′ is explained here. In the IR spectrum (KBr) of 3c′, the broad band at 3227 cm−1 corresponds to hydroxyl stretching and the bands appearing at 2978 and 2850 cm−1 correspond to aliphatic and aromatic stretchings respectively. The bands appearing at 2737 and 2677 cm−1 represent the secondary amine salt (NH2)+. The sharp band at 1674 cm−1 indicates the carbonyl stretching and the bands at 1585 and 1437 cm−1 correspond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of the benzenoid rings. The bands at 1338 and 1221 cm−1 confirm the presence of –C–N and C–O groups respectively. The characteristic band appears at 982, 833, 760, and 696 cm−1 representing mono- and di-substituted benzene rings of 3c′. In the 1H-NMR spectrum of 3c′, the doublet signal at δH 1.03 ppm (J = 6.7 Hz) indicates the presence of three protons of aliphatic CH3 group and the doublet of doublet signal at δH 3.41 ppm (J = 15.7 and 8.8 Hz) shows the presence of one proton of –CH group adjacent to amino group. The two triplet signals at δH 3.47 ppm (J = 6.4 Hz) and 3.68 ppm (J = 7.0 Hz) confirm the presence of four protons of two –CH2 groups. A broad singlet signal at δH 5.31 ppm indicates one proton of the hydroxyl group and doublet signal at δH 6.24 ppm (J = 4.1 Hz) indicates one proton of –CH attached to hydroxyl function. Multiplet signals appear in between δH 7.33 and 8.07 ppm showing the presence of nine aromatic protons of two phenyl rings and a broad singlet signal at δH 9.12 ppm indicating two protons of ammonium group of compound 3c′. In the 13C-NMR spectrum of 3c′, the signal at δC 9.79 ppm indicates the carbon of the aliphatic methyl group; the signal at δC 35.28 confirms the CH2 adjacent to carbonyl group; the signals at 40.20 and 58.72 ppm show two carbons (–CH and –CH2) attached to ammonium group and another signal at δC 58.72 ppm indicates the carbon of –CH2 group attached to ketone function. A signal at δC 69.96 ppm confirms the carbon of –CH group attached to hydroxyl group. The eight signals appear in-between δC 126.35 and 141.67 ppm elucidating the eight symmetric carbons of two phenyl rings; a signal appears at δC 196.56 ppm confirming the carbon of keto function of 3c′. Two distinguishing isotopic peaks observed at m/z 318 and 320 in the ratio of 3:1 in the mass spectrum of 3c′ confirm the protonated molecular ion [M + H]+ of compound 3c′. Single crystal X-ray diffraction study unequivocally confirmed the structure of 3c′ deduced from above spectral techniques as well as its stereo chemical aspects (Fig. 3). The crystallographic data for compound 3c′ has been deposited with the Cambridge Crystallographic Data Centre (CCDC) as supplementary publication number CCDC 1443803.40
C stretching of the benzenoid rings. The bands at 1338 and 1221 cm−1 confirm the presence of –C–N and C–O groups respectively. The characteristic band appears at 982, 833, 760, and 696 cm−1 representing mono- and di-substituted benzene rings of 3c′. In the 1H-NMR spectrum of 3c′, the doublet signal at δH 1.03 ppm (J = 6.7 Hz) indicates the presence of three protons of aliphatic CH3 group and the doublet of doublet signal at δH 3.41 ppm (J = 15.7 and 8.8 Hz) shows the presence of one proton of –CH group adjacent to amino group. The two triplet signals at δH 3.47 ppm (J = 6.4 Hz) and 3.68 ppm (J = 7.0 Hz) confirm the presence of four protons of two –CH2 groups. A broad singlet signal at δH 5.31 ppm indicates one proton of the hydroxyl group and doublet signal at δH 6.24 ppm (J = 4.1 Hz) indicates one proton of –CH attached to hydroxyl function. Multiplet signals appear in between δH 7.33 and 8.07 ppm showing the presence of nine aromatic protons of two phenyl rings and a broad singlet signal at δH 9.12 ppm indicating two protons of ammonium group of compound 3c′. In the 13C-NMR spectrum of 3c′, the signal at δC 9.79 ppm indicates the carbon of the aliphatic methyl group; the signal at δC 35.28 confirms the CH2 adjacent to carbonyl group; the signals at 40.20 and 58.72 ppm show two carbons (–CH and –CH2) attached to ammonium group and another signal at δC 58.72 ppm indicates the carbon of –CH2 group attached to ketone function. A signal at δC 69.96 ppm confirms the carbon of –CH group attached to hydroxyl group. The eight signals appear in-between δC 126.35 and 141.67 ppm elucidating the eight symmetric carbons of two phenyl rings; a signal appears at δC 196.56 ppm confirming the carbon of keto function of 3c′. Two distinguishing isotopic peaks observed at m/z 318 and 320 in the ratio of 3:1 in the mass spectrum of 3c′ confirm the protonated molecular ion [M + H]+ of compound 3c′. Single crystal X-ray diffraction study unequivocally confirmed the structure of 3c′ deduced from above spectral techniques as well as its stereo chemical aspects (Fig. 3). The crystallographic data for compound 3c′ has been deposited with the Cambridge Crystallographic Data Centre (CCDC) as supplementary publication number CCDC 1443803.40
 |
| | Fig. 2 Structures of all synthesized compounds. | |
 |
| | Fig. 3 ORTEP diagram of compound 3c′. | |
Enantiomeric nature of the synthesized Mannich derivatives
The Mannich derivatives (3a–g and 3a′–g′) were synthesized from enantiomeric amines Ψ-PPA 1 and 1′ and obtained Mannich derivatives were expected to be enantiomers. The obtained Mannich derivatives 3a–g and 3a′–g′ were analyzed by ordinary and chiral HPLC. Ordinary HPLC analysis showed same retention time (RT) for 3a–g and 3a′–g′ whereas chiral HPLC analysis showed two different RT's for 3a–g and 3a′–g′ respectively. To confirm the enantiomeric nature of the synthesized compounds, racemic Mannich derivatives were prepared using dl-Ψ-PPA and the obtained RT's from chiral HPLC were compared with the RT's of 3a–g and 3a′–g′. For example, Mannich derivative of dl-Ψ-PPA using paraformaldehyde and 2,4-dichloroacetophenone was prepared by refluxing in ethyl alcohol with aqueous hydrochloric acid as per the conventional method reported in the literature,16 analyzed by ordinary/chiral HPLC and measured the SOR. A single peak observed in ordinary HPLC and SOR was found to be as +0.12° for the prepared racemic Mannich derivative. Two different peaks with equal intensity were found at RT's 7.24 and 8.41 min by chiral HPLC. These RT's were matches with compound 3d (RT 7.30 min) and 3d′ (RT 8.44 min) which were synthesized from 1 and 1′ respectively. Hence, the prepared Mannich derivatives 3a–g and 3a′–g′ are confirmed as enantiomerically pure (ee 99.9%).
Anti-proliferative effect of Mannich derivatives
The anti-proliferative effect of Mannich derivatives (3a–g and 3a′–g′) were examined against A549 and HepG-2 cells using MTS analysis. The anti-proliferative results were observed at 150 to 75 μM in A549 cells and 150 to 25 μM in HepG-2 cells, respectively. Interestingly, among all the tested compounds, 3a, 3f, 3a′, 3d′, 3e′ and 3f′ showed good anti-proliferative activity against both the A549 and HepG-2 cells compared to other compounds (Fig. 4). Whereas compounds 3a, 3d, 3a′, 3e′, 3f′ and 3g′ exhibited much better anti-proliferative activity against A549 cells with IC50 value of 100 μM. In particular, 3d′ showed potent activity against A549 cells with IC50 value of 75 μM. Among the tested compounds, 3a–b, 3e–f, 3a′ and 3d′–f′ showed good anti-proliferative activity against HepG-2 cells. Especially, compounds 3b, 3d′ and 3f′ revealed much better anti-proliferative activity against HepG-2 cells with IC50 value of 75 μM. Interestingly, 3a exhibited maximum anti-proliferative activity against HepG-2 cells with IC50 value of 25 μM. However, the compounds 3e–f, 3a′–b′, 3e′ and 3g′ emerged as the least active and less attractive series against HepG-2 cells exhibiting IC50 values of 100 μM. All concentrations used in the experiment decreased the cell viability significantly (P < 0.05) in a concentration dependent manner. Interestingly, we found that compound 3a showed no toxicity against Vero cells (normal kidney epithelial cells originated from Cercopithecus aethiops) up to 600 μM. The structures of the synthesized Mannich derivatives 3a–g and 3a′–g′ with respect to their substitutions were correlated against anti-proliferative activities with A549 and HepG-2 cells. It was noticed that unsubstituted (3a & 3a′), o,p-dichloro substituted (3d), p-hydroxy substituted (3e′), m-nitro substituted (3f′) and p-methoxy substituted (3g′) Mannich derivatives showed better anti-proliferative activity against A549 cells. Mannich derivatives with p-methyl (3b & 3b′) and p-chloro (3c & 3c′) substitutions showed least activity. When comparing the stereo configuration of Mannich derivatives with anti-proliferative activity against A549 cells, it was noticed that (1R,2R)-Mannich derivatives 3a′–g′ showed better activity than (1S,2S)-Mannich derivatives 3a–g. In the case of HepG-2 cells unsubstituted (3a & 3a′), p-methyl (3b), o,p-dichloro substituted (3d′), p-hydroxy substituted (3e & 3e′) and m-nitro substituted (3f & 3f′) Mannich derivatives showed good anti-proliferative activities. p-Methyl (3b), o,p-dichloro (3d′) and m-nitro (3f′) substituted Mannich derivatives exhibited better anti-proliferative activity against HepG-2 cells with IC50 value of 75 μM. Interestingly, as in A549 cells, here also compound 3a which had no substitutions showed potential anti-proliferative activity at 25 μM concentration. When comparing the stereo configuration of Mannich derivatives with anti-proliferative activity against HepG-2 cells, it was noticed that both (1R,2R)- and (1S,2S)-Mannich derivatives showed better activities. When comparing the anti-proliferative activity of compounds against two tested cells, it was noticed that maximum compounds containing electron donating groups at o-, p-position and electron withdrawing group at m-position showed good activity. The better activity of (1R,2R)-Mannich derivatives than (1S,2S)-isomers indicated that the target receptor in both tested cells bound with more affinity.
 |
| | Fig. 4 Comparison of anticancer activity of synthesized Mannich derivatives (3a–g and 3a′–g′) against A549 and HepG-2 cells. DMSO was used as a vehicle control. Data are mean ± SD of three independent experiments with each experiment conducted in triplicate. Positive control: HepG-2 cells-doxorubicin 8 ± 0.23 μM (IC50) and A549 cells-cisplatin 9.80 ± 0.41 μM (IC50). | |
Immunofluorescence and colony formation studies
HepG-2 cells were treated with 3a and subjected to confocal microscopic studies. Normally, confocal microscopic image shows the apoptosis or morphological changes using different fluorescent dyes. In this study, we used DAPI, FITC and PI fluorescence stains. DAPI binds strongly to A-T rich regions in DNA and passes through an intact cell membrane. FITC acts as a phosphatidyl serine tracer and suggests the presence of apoptosis. PI can only penetrate cells where the cell membrane has been compromised. Our results showed that significant morphological changes were found after treatment with 3a in HepG-2 cells in confocal microscopic images. FITC stain showed membrane level changes; DAPI and PI stain showed fragmentation and condensation of nuclei (Fig. 5). To determine the potential anticancer effect of 3a in HepG-2 cells, in vitro colony formation assays were undertaken. A549 cells were treated with 3a at concentrations of 25, 50 and 75 μM for 10 days. There was dramatic increase in the colony forming ability when compared to the control (Fig. 6).
 |
| | Fig. 5 Fluorescence staining for the detection of apoptosis in HepG-2 cells. Cells were treated with 25, 50 and 75 μM concentration of 3a for 24 h. The fluorescent signals of DAPI, FITC and PI were examined under confocal laser scanning microscope, Zeiss, 2009. DAPI (blue color), FITC (green color), PI (red color) and merged image. HepG-2 control cells (a1-DAPI, a2-FITC, a3-PI & a4-merged), treated cells (25 μM: b1-DAPI, b2-FITC, b3-PI & b4-merged), treated cells (50 μM: c1-DAPI, c2-FITC, c3-PI and c4-merged) and treated cells (75 μM: d1-DAPI, d2-FITC, d3-PI and d4-merged). Arrows indicate apoptotic cancer cells. | |
 |
| | Fig. 6 Effect of colony formation assay was examined against HepG-2 cells using compound 3a. HepG-2 cells were treated with or without compound 3a and number of colonies were counted 10th day later after cells were stained with crystal violet. Data shown are representatives of three separate experiments. | |
Apoptosis analysis
The flow cytometry is one of the efficient and specific methods to investigate the cell death analysis. The fractions of cell populations in different quadrants are analyzed using quadrant statistics such as lower left quadrant (R2-living cells), lower right quadrant (R3-early apoptosis cells), the upper right quadrant (R4-the late apoptosis cells) and the upper left quadrant (R5-dead cells). To examine the apoptosis effect of 3a, we treated HepG-2 cells with 25, 50, 75 and 100 μM concentrations of 3a for 24 h. Apoptotic cells were measured by annexin V-FITC/PI staining assay as per manufacturer's guidelines. The results showed that after treatment with 3a for 24 h there was dramatic increase in the apoptosis effect on HepG-2 cells when compared to the control cells (Fig. 7A–F). Interestingly, when increasing the concentrations, early (R3) and late (R4) apoptotic cells gradually increased compared to the control cells. Our results revealed that the population of the living cells (R2) decreased and the percentage of early (R3) and late (R4) apoptotic cells increased.
 |
| | Fig. 7 HepG-2 cells were treated with 25, 50, 75 and 100 μM concentrations of 3a for 24 h and apoptosis was analyzed by flow cytometry using annexin V-FITC/PI detection kit. The results showed that 3a significantly increased the apoptosis in a dose dependent manner. Annexin V-positive cells were considered as apoptotic cells. Data are mean ± SD of three independent experiments with each experiment conducted in triplicate. R2 = live cells; R3 = Early apoptotic cells; R4 = Late apoptotic cells; R5 = dead cells. | |
Cell cycle arrest
Overgrowth of cancer is due to dysfunction in the regulation of the cell cycle that appears in growth of cells, although cancer progression can be strongly limited by conquest of the cell cycle.23 Based on the inhibitory effect on cell proliferation, we also examined the effects of 3a on cell cycle progression. We investigated whether 3a treatment affected the cell cycle arrest in HepG-2 cells. Treated and untreated HepG-2 cells were subjected to cell cycle arrest analysis using flow cytometry. We used different concentrations of 3a (25, 50, 75 and 100 μM) for 24 h treatment. Our results indicated that after treatment with 3a, cell cycle arrest in G1 phase gradually decreased whereas G2 phase was increased when compared to the control. The percentage of cells in G0/G1 in control vs. 3a in different concentrations in HepG-2 cells were 74.95% vs. 69.15% at 25 μM, 74.95% vs. 59.86% at 50 μM, 74.95% vs. 47.38% at 75 μM and 74.95 vs. 37.62% at 100 μM, respectively (Fig. 8A–F). Compared with the condition of control cells, the proportion of cells at S phase significantly decreased in HepG-2 cells in the presence of 3a. In addition, 3a induced cell accumulation in the G2/M phase, which represents a cell cycle arrest at the G2/M phase, while G0/G1 phase accumulation was induced in HepG-2 cells, reflecting a G0/G1 cycle arrest.
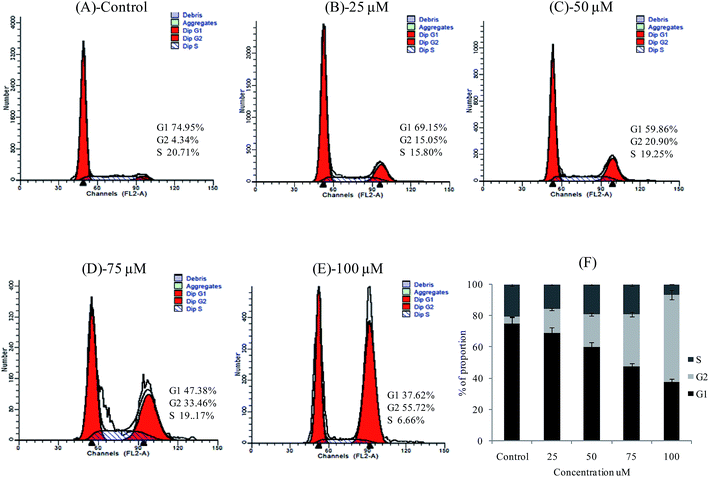 |
| | Fig. 8 Cell cycle arrest in HepG-2 cells treated with 3a (25, 50, 75 and 100 μM). Representative results showing the percentage of cells in G1, G2 and S phase in HepG-2 cells without or with 3a treatment for 24 h as detected by flow cytometry. Data are mean ± SD of three independent experiments with each experiment conducted in triplicate. | |
Expression of mitochondrial signaling pathway
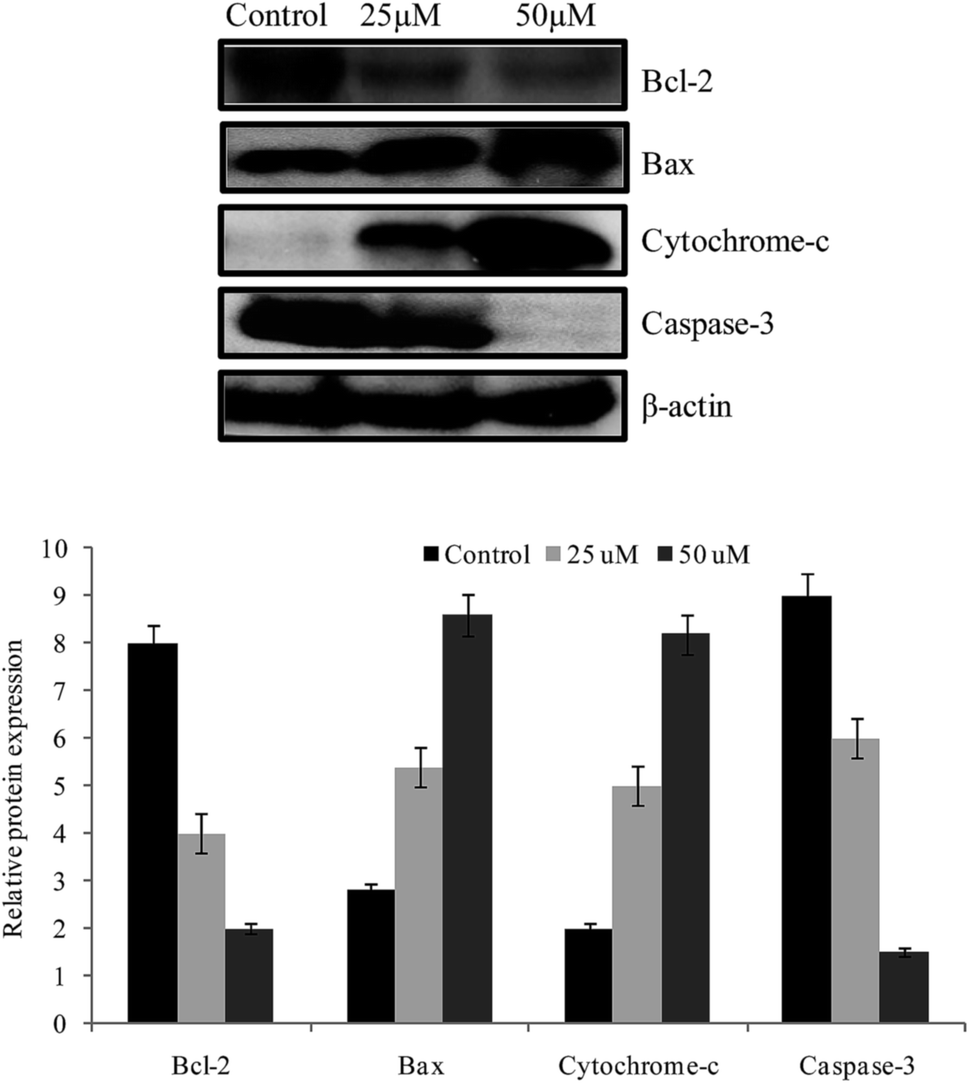
We examined the expressions of Bcl-2, Bax, cytochrome c and caspase-3 proteins by western blotting after 24 h treatment of 3a. Bcl-2 is localized in the outer mitochondrial membrane and involved in over expressions during all cancer. Our results revealed that Bcl-2 expression was down regulated and Bax expression was increased after treatment with 3a against HepG-2 cells at 25 and 50 μM concentrations after 24 h (Fig. 9). The intrinsic apoptosis signal pathway is the release of cytochrome c in mitochondria and the extrinsic pathway is the activation of death receptors.41 Besides, we evaluated the expression of apoptosis involving cytochrome c after treatment with 3a against HepG-2 cells. Our results showed that the level of cytochrome c was increased after treatment with 3a (Fig. 9). After expression, cytochrome c binds with caspase-9 and activates caspase-3 to promote apoptosis. Our results indicated that the cleavage of caspase-3 was up regulated by a cytochrome c release mediated pathway (Fig. 9). Finally, after treatment of 3a apoptosis was induced through caspase-3 mediated pathway.
 |
| | Fig. 9 Expression of Bcl-2, Bax, cytochrome c and caspase-3 cells was measured following exposure to 3a for 24 h. Western blotting analysis of Bcl-2, Bax, cytochrome c and caspase-3 after 3a treatment at different concentrations for 24 h. Western blotting was performed using suitable antibodies. β-Actin was used as an internal control. P < 0.05 compared to 3a treated HepG-2 cells. | |
JAK2/STAT3 expression
JAK/STAT is an important therapeutic target in cancer treatment. JAK/STAT is a key oncogenic signaling cascade involved in tumorigenesis and is found commonly involved in many types of cancers.42 Furthermore, the activation of JAK/STAT expression controls the major cellular responses.43 Beside, down regulation of JAK/STAT activity leads to induction of an apoptotic response. Moreover, the inhibition of STAT3 phosphorylation is majorly achieved via the inhibition of upstream STAT3 activating receptor tyrosine kinases such as JAK1 and JAK2.44 In the present study, we investigated the underlying mechanism of action of 3a and its effects on JAK2/STAT3 signaling in HepG-2 cells. The results showed that phosphorylation of JAK2 and STAT3 were significantly inhibited in a dose dependent manner after 24 h treatment with 3a in HepG-2 cells (Fig. 10A). Next, we examined the time dependent inhibition of JAK2/STAT3. HepG-2 cells were treated with 3a for 6 h and 12 h and cells were lysed for western blot analysis. Compound 3a induced a notable reduction in the levels of p-STAT3 and p-JAK2 after a short time, with apparent p-STAT3 and p-JAK2 inhibition occurring at around 6 h and 12 h (Fig. 10B), and 3a decreased p-STAT3 and p-JAK2 in a time dependent manner. The above results suggested that the JAK family kinases are potential target of 3a, which induced apoptosis in HepG-2 cells through inhibition of the JAK2/STAT3 signaling pathway. Finally, we confirmed that apoptosis mechanism was involved in JAK2/STAT3 mediated signaling pathway in HepG-2 cells. A proposed mechanism is as follows: JAK/STAT inhibition after treatment with 3a in HepG-2 cells transmits the chemical signals from outside of the cell through cytokine. After JAK is activated, STAT3 protein binds to the phosphorylated receptor, translocated into the cell nucleus, binds to DNA and promotes transcription of genes responsive to STAT. Then, STAT3 connected to anti-apoptotic gene Bcl-2 and Bcl-2 is activated. The down regulation of Bcl-2 is connected to the expression of pro-apoptosis Bax gene. Bax can permeabilize the mitochondrial outer membrane, releasing cytochrome c and activate the apoptosis involving caspases. Finally, caspase proteins trigger the cell death or apoptosis.
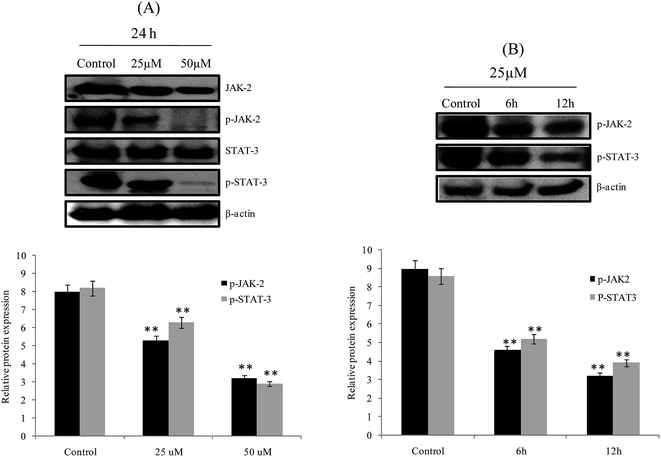 |
| | Fig. 10 Compound 3a inhibited constitutive JAK2/STAT3 signaling pathway in HepG-2 cells in a time and dose dependent manner. HepG-2 cells were treated with 3a (A) for 24 h (25 and 50 μM), (B) 12 h and 6 h (25 μM). Then treated and untreated HepG-2 cells were lysed for western blot analysis. β-Actin was used as a loading control. **P < 0.05 compared to 3a treated HepG-2 cells. | |
In vivo model
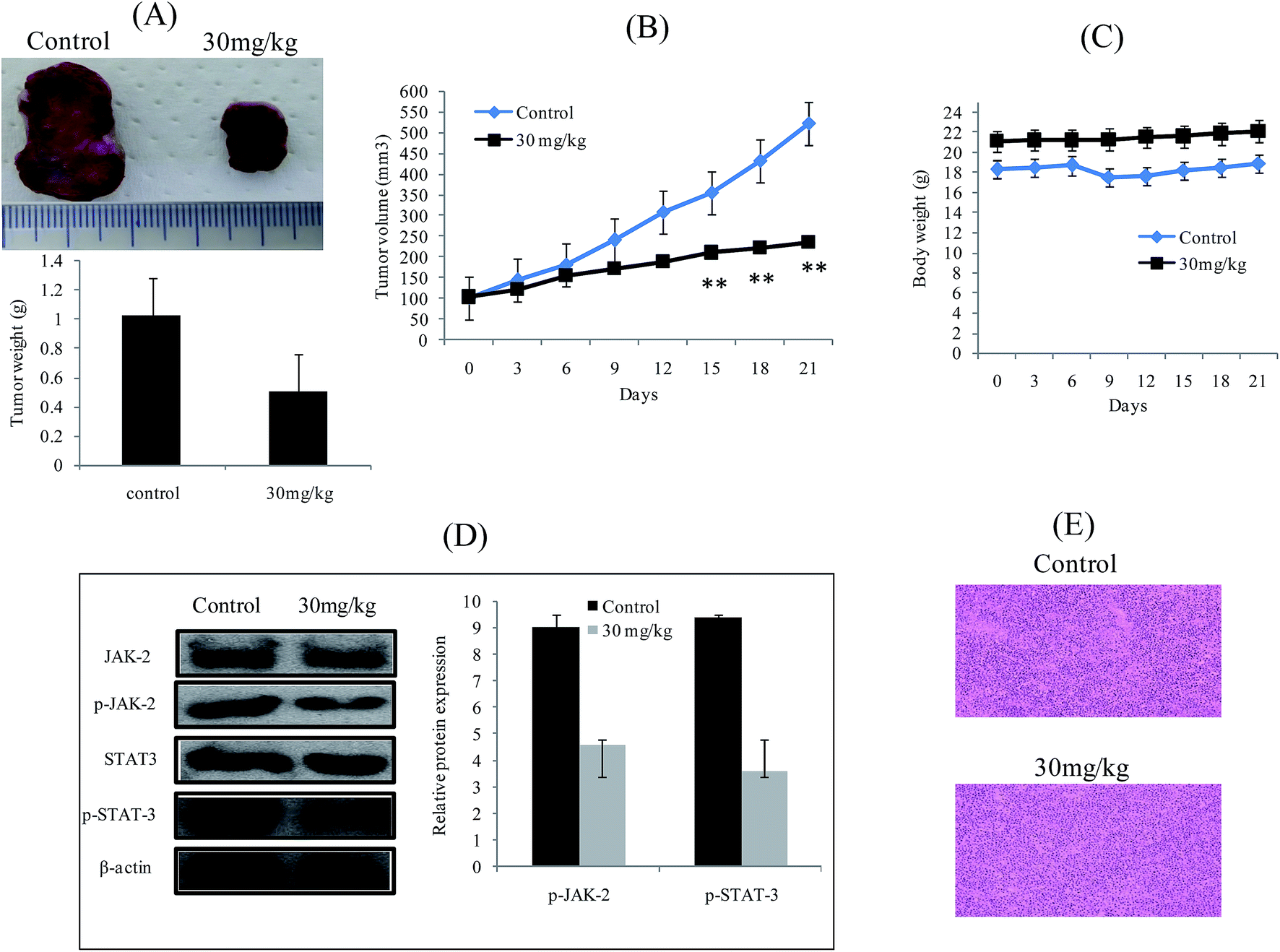
We evaluated the anticancer effect of 3a in a xenograft tumor model by subcutaneously injecting the HepG-2 cells. On the 7th day after implantation, when tumors reached 100 mm3 volume, mice were randomly divided into two groups such as control and treated (30 mg kg−1). Tumor size and body weight were measured for each animal from the beginning of 3a treatment to the 21st day. The average tumor weight was significantly reduced in the 3a treated mice compared with the control mice (Fig. 11A and B). Interestingly, 3a did not affect the overall body weight during the experimental period, suggesting no apparent toxicity (Fig. 11C). Western blot analysis showed that the phosphorylation of JAK2 and STAT3 were also substantially reduced in 3a treated tumors than in control tumors (Fig. 11D). Histochemical analysis showed detection of apoptotic cells after treatment of 3a (Fig. 11E). These data suggest that 3a significantly suppressed tumor growth in animals through the blockage of JAK2/STAT3 signaling pathway.
 |
| | Fig. 11 Effect of 3a on tumor growth in xenograft nude mice. (A) BALB/c nude mice were subcutaneously injected with HepG-2 cells and intraperitoneally administered with 30 mg kg−1 of 3a for 21 days (every 3 days) and average weight of the tumors from mice was compared with control. (B) Tumor volumes were measured with a calliper every 3rd day. Significant difference was compared with the control group (n = 4) and **p < 0.01. (C) The body weight changes were monitored during the test period. (D) Western blot analysis for protein expression. (E) H & E staining for detection of apoptosis cells. | |
Molecular docking studies
Docking method was validated using crystallized and docked ligand with JAK-2 receptor 2XA4 (Fig. 12A1 and A2). Compound 3a showed the binding energy value of −6.10 kcal mol−1 with the docked JAK-2 receptor and it interacted with 22 active sites of amino acids namely Leu-855, Gly-856, Lys-857, Val-863, Ala-880, Val-881, Lys-882, Val-911, Met-929, Glu-930, Tyr-931, Leu-932, Pro-933, Tyr-934, Gly-935, Ser-936, Arg-980, Asn-981, Ile-982, Leu-983, Gly-993 and Asp-994 with two hydrogen bonds. In 3a, O–H interacted with the N–H of Ser-936 and formed a hydrogen bond with the bond length of 2.8 Å. Also, CO interacted with the N–H of Leu-932 and formed a hydrogen bond with the bond length of 1.8 Å. In addition to the hydrogen bonds, hydrophobic interaction was exhibited between phenyl ring of the 3a and Leu-855, Ala-880, Val-911, Leu-932, Leu-983 and Gly-993 amino acids. Binding interaction of the 3a with the JAK-2 receptor was showed in Fig. 12B1 and B2. Compound 3a showed the binding energy value of −6.04 kcal mol−1 with the docked Bcl-2 receptor and it interacted with the 14 active sites of amino acids namely Phe-63, Tyr-67, Asp-70, Phe-71, Met-74, Val-92, Leu-96, Gly-104, Arg-105, Ala-108, Glu-111, Phe-112 and Val-115. In 3a, O–H interacted with the CO of Ala-108 and formed a hydrogen bond with the bond length of 1.9 Å. Furthermore, π–π interaction was exhibited between phenyl rings of the 3a and Phe-63, Tyr-67, Phe-71 and Phe-112 amino acids. In addition to the hydrogen bond and π–π interactions, hydrophobic interaction was exhibited between phenyl ring of the compound with Leu-96, Gly-104, Ala-108 and Val-115 amino acids. Binding interaction of the 3a with the Bcl-2 receptor was showed in Fig. 12C1 and C2.
 |
| | Fig. 12 Molecular docking results of 3a with JAK-2 and Bcl-2 receptors: A1 & A2-method validation using crystallized and docked ligand with JAK-2 receptor; B1 & B2-docking pose of 3a with JAK-2; C1 & C2-docking pose of 3a with Bcl-2. | |
Conclusion
In summary, we have described the synthesis of seven enantiomeric pairs of Mannich derivatives 3a–g and 3a′–g′ from Ψ-PPA 1 and 1′ respectively using recyclable H2SO4·SiO2 catalyst. All the synthesized compounds were found to be chirally pure by chiral HPLC analysis and the structures of all compounds were well characterized with the help of spectral techniques. The obtained compounds were proved as potential in vitro and in vivo anticancer agents. All compounds were found to have potential anti-proliferative activities against A549 and HepG-2 cells. Among the tested compounds, 3a, 3f, 3a′, 3d′, 3e′ and 3f′ exhibited promising anti-proliferative activity against A549 and HepG-2 cells at 150 to 25 μM concentrations. In particular, compound 3a showed promising activity at 25 μM against HepG-2 cells and compound 3d′ showed potential activity with IC50 value at 75 μM against A549 cells. Furthermore, 3a exhibited potent apoptosis properties against HepG-2 cells, dysregulation of mitochondrial functions, cell cycle arrest at G2/M phase and JAK2/STAT3 inhibition. Importantly, 3a significantly suppressed HepG-2 tumorigenesis in in vivo model. The molecular docking studies showed good binding energy values. These results suggest that 3a is a novel blocker of JAK2/STAT3 signaling pathway and exerts both anti-proliferative and apoptotic activities in HepG-2 cells in vitro and in vivo.
Materials and methods
Reagents and chemicals
Enantiomers of Ψ-PPA were obtained from Malladi Drugs & Pharmaceuticals Limited, Chennai. All other reagents and solvents used were obtained from commercial sources and used as such. Analytical thin layer chromatography (TLC) was performed on pre-coated aluminium sheets of silica gel G/UV-254 nm of 0.2 mm thickness (Merck, Germany). DMEM, PBS, TRIZOL reagent, FITC, PI, DAPI and DMSO were obtained from Sigma. Fetal Bovine Serum (FBS) was obtained from Biowest (France). CellTiter 96® AQueous one solution was purchased from Promega (G3580), USA. Annexin V-FITC/PI detection kit was obtained from Biolegend. CycleTEST™ PLUS DNA reagent kit was purchased from BD Biosciences, California. Bax, Bcl-2, caspase-3 and cytochrome c were purchased from BD Biosciences. JAK-2, p-JAK-2, STAT3, p-STAT3, β-actin and horseradish peroxidise conjugate were obtained from cell signaling technology. Chemiluminescence Luminol Reagent kit was purchased from Santa Cruz Biotechnology, USA.
Typical experimental procedure to synthesize Mannich derivatives 3a–g and 3a′–g′
Under nitrogen atmosphere, (1S,2S)-Ψ-PPA 1 (10.0 g, 66 mmol), isopropyl alcohol (70 mL), paraformaldehyde (2.2 g, 73 mmol) and 10% H2SO4·SiO2 (10 mol%, 6.5 g) were mixed and stirred for 10 min at ambient temperature followed by addition of 70 mmol of substituted acetophenone 2a–g. It was slowly heated and the reaction mass was maintained at 45–50 °C for about 6–10 h. Completion of reaction was confirmed by TLC (10% methanol in dichloromethane). Catalyst separated by filtration and isopropyl alcohol was distilled off from the filtrate using rotary evaporator under vacuum. The obtained residue was dissolved in dichloromethane (50 mL), washed with 5% sodium bicarbonate solution. The organic layer was acidified using ethanolic hydrochloric acid at below 10 °C and the obtained precipitate was filtered. Further, it was recrystallized in ethanol to get white shiny crystals of hydrochloride salt of Mannich derivatives 3a–g in 86 to 95% yield. The same method was repeated with (1R,2R)-Ψ-PPA 1′ instead of 1 to get white glittering crystals of hydrochloride salts of Mannich derivatives 3a′–g′ in 87 to 94% yield.
Preparation of sulphuric acid immobilized on silica gel catalyst. Slowly, sulphuric acid (10 g) was added to the suspension of silica gel (100 g) in methanol (300 mL). The contents were heated to 60–65 °C and maintained for an hour. Methanol was completely distilled off from the mass and the obtained sulphuric acid was immobilized on silica gel catalyst and was further dried at 60 °C to get a free flowing powder. Catalyst was preserved in nitrogen atmosphere.
Recycle of used catalyst. The recovered catalyst from the Mannich reaction was reused (3 to 5 times) after washing with methanol and drying at 80 °C for 6 h.
N-((1S,2S)-1-Hydroxy-1-phenylpropan-2-yl)-3-oxo-3-phenylpropan-1-amine HCl (3a). White crystalline powder; yield 94%; mp: 107–110 °C; [α]25D: +58.5° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 6.94 min); IR (KBr, cm−1): 3350 (broad, OH), 2762 and 2640 (NH2+), 1682 (CO), 1599 and 1448 (benzenoid), 1321 (C–N), 1211 (C–O), 746 and 704 (mono substituted benzene rings); 1H-NMR (DMSO-d6, 400 MHz): δH (ppm) 0.95 (d, J = 6.6 Hz, 3H, CH3), 3.14 (t, 1H, J = 6.8 Hz, N–CH), 3.58–3.60 (m, 4H, CH2–CH2), 5.21 (bs, 1H, OH), 6.14 (d, 1H, J = 4.2 Hz, O–CH), 7.33–7.98 (m, 10H, aromatic), 8.91 (bs, 2H, NH2+); 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 9.84 (CH3), 35.19 (N–CH), 39.78 (N–CH2) 58.66 (Ph–CH2), 70.01 (O–CH), 126.31, 127.78, 128.43, 128.62, 129.36, 134.16, 136.46, 141.59 (aromatic carbons) 197.51 (CO); MS-ESI: m/z at 284.1 [M + H]+; anal. calcd for C18H22ClNO2: C, 67.60; H, 6.93; N, 4.38. Found C, 67.58; H, 6.95; N, 4.42.
N-((1S,2S)-1-Hydroxy-1-phenylpropan-2-yl)-3-oxo-3-(p-tolyl)propan-1-amine HCl (3b). White crystalline powder; yield 90%; mp: 204–206 °C; [α]25D: +47.1° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 11.45 min); IR (KBr, cm−1): 3339 (broad, OH), 2978 and 2921 (NH2+), 1677 (CO), 1607 and 1573 (benzenoid bands), 1359 (C–N), 1222 (C–O), 817 (o,p-di substituted benzene ring) and 704 (mono substituted benzene rings); 1H-NMR (DMSO-d6, 400 MHz): δH (ppm) 0.97 (d, J = 6.7 Hz, 3H, CH3), 2.39 (s, 3H, Ph–CH3), 3.42 (t, J = 4 Hz, 2H, CO–CH2), 3.51 (1H, m, N–CH), 3.56 (t, J = 7.3 Hz, 2H, N–CH2), 5.21 (bs, 1H, OH), 6.22 (d, J = 4.0 Hz, 1H, O–CH), 7.29–7.91 (m, 9H, aromatic) and 8.83 (bs, 2H, NH2+), 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 9.83 (–CH3), 21.66 (Ph–CH3), 35.01 (CO–CH2), 40.42 (N–CH2), 58.74 (N–CH), 70.06 (O–CH), 126.30, 127.87, 128.57, 128.68, 129.90, 133.94, 141.42, 144.76 (aromatic carbons) and 197.11 (CO); MS-ESI: m/z at 298.1 [M + H]+; anal. calcd for C19H24ClNO2: C, 68.35; H, 7.25; N, 4.20. Found C, 68.48; H, 7.30; N, 4.21.
3-(4-Chlorophenyl)-N-((1S,2S)-1-hydroxy-1-phenylpropan-2-yl)-3-oxopropan-1-amine HCl (3c). White crystalline powder; yield 88%; mp: 138–141 °C; [α]25D: +38.8° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 7.35 min); IR (KBr, cm−1): 3343 (b, OH), 2972 and 2816 (NH2+), 1686 (CO), 1588 and 1574 (benzenoid), 1315 (C–N), 1215 (C–O), 783 (o,p-di substituted benzene ring) and 698 (mono substituted benzene ring); 1H-NMR (DMSO-d6, 400 MHz): δH (ppm) 1.00 (d, J = 6.7 Hz, 3H, CH3), 3.41 (dd, J = 15.7 & 8.8 Hz, 1H, N–CH), 3.45 (t, J = 5.6 Hz, 2H, CO–CH2), 3.65 (t, J = 7.0 Hz, 2H, N–CH2), 5.28 (bs, 1H, OH), 6.22 (d, J = 4.1 Hz, 1H, O–CH), 7.31–8.05 (m, 9H, aromatic), 9.05 (bs, 2H, NH2+), 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 9.79 (CH3), 35.25 (CO–CH2), 40.16 (N–CH), 58.65 (N–CH2), 69.96 (O–CH), 126.34, 127.80, 128.65, 129.50, 130.40, 135.19, 139.05, 141.64 (aromatic carbons), 196.54 (CO); MS-ESI: m/z [M + H]+ at 318 & 320 in the ratio of 3:1 respectively; anal. calcd for C18H21Cl2NO2: C, 61.02; H, 5.97; N, 3.95. Found C, 61.15; H, 6.00; N, 3.95.
3-(2,4-Dichlorophenyl)-N-((1S,2S)-1-hydroxy-1-phenylpropan-2-yl)-3-oxopropan-1-amine HCl (3d). White crystalline powder; yield 95%; mp: 198–200 °C; [α]25D: +27.8° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 7.30 min); IR (KBr, cm−1): 3337 (b, OH), 2978 and 2826 (NH2+), 1699 (CO), 1583 and 1551 (benzenoid), 1314 (C–N), 1203 (C–O), 980 (tri substituted benzene ring), 741 (mono substituted benzene ring); 1H-NMR (DMSO-d6, 400 MHz): δH (ppm) 0.97 (d, J = 6.7 Hz, 3H, CH3); 3.40 (t, J = 6.8 Hz, 2H, CO–CH2), 3.56 (s, 1H, N–CH), 3.59 (t, J = 7.3 Hz, 2H, N–CH2), 5.25 (bs, 1H, OH), 6.19 (d, J = 4 Hz, 1H, O–CH), 7.36–7.86 (m, 8H, aromatic) and 9.09 (bs, 2H, NH2+); 13C-NMR (DMSO-d6, 400 MHz): δC (ppm) 9.69 (CH3), 52.21 (Ph–CH2), 58.31 (N–CH), 69.67 (N–CH2), 71.81 (O–CH), 126.23, 127.71, 128.18, 128.57, 130.11, 130.67, 131.74, 136.30, 137.19, 141.42 (aromatic carbons) and 197.99 (CO); MS-ESI: m/z [M + H]+ at 352, 354 & 356 in the ratio of 9:6:1 respectively; anal. calcd for C18H20Cl3NO2: C, 55.62; H, 5.19; N, 3.60. Found C, 55.75; H, 5.24; N, 3.63.
N-((1S,2S)-1-Hydroxy-1-phenylpropan-2-yl)-3-(3-hydroxyphenyl)-3-oxopropan-1-amine HCl (3e). White crystalline powder; yield 89%; mp: 172–175 °C; [α]25D: +57.6° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 4.52 min); IR (KBr, cm−1): 3371 (b, OH), 2991 and 2799 (NH2+), 1681 (CO), 1604 and 1583 (benzenoid), 1316 (C–N), 1262 (C–O), 995 (tri substituted benzene ring) and 703 (mono substituted benzene ring); 1H-NMR (DMSO-d6, 400 MHz): δH (ppm) 1.05 (d, J = 6.6 Hz, 3H, CH3); 3.35 (s, 1H, N–CH), 3.43 (t, J = 5.9 Hz, 2H, CO–CH2), 3.55 (m, 2H, N–CH2); 4.61 (bs, 1H, C–OH), 6.48 (d, J = 3.2 Hz, 1H, O–CH), 7.13–7.48 (m, 9H, aromatic), 8.44 and 9.08 (two bs, 2H, NH2+); 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 12.83 (CH3), 34.96 (N–CH), 40.22 (CO–CH2), 58.99 (N–CH2), 73.99 (O–CH), 114.57, 119.37, 121.28, 127.64, 128.69, 128.92, 130.41, 137.66, 141.52, 158.09 (aromatic carbons) and 197.83 (CO); MS-ESI: m/z at 300 [M + H]+; anal. calcd for C18H22ClNO3: C, 64.38; H, 6.60; N, 4.17. Found C, 64.48; H, 6.65; N, 4.20.
N-((1S,2S)-1-Hydroxy-1-phenylpropan-2-yl)-3-(3-nitrophenyl)-3-oxopropan-1-amine HCl (3f). White crystalline powder; yield 92%; mp: 158–159 °C; [α]25D: +56.9° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 5.27 min); IR (KBr, cm−1): 3356 (b, OH), 2924 and 2853 (NH2+), 1668 (CO), 1602 and 1520 (benzenoid), 1562 and 1393 (NO2), 1252 (C–N), 1219 (C–O), 852 (di substituted benzene ring) and 702 (mono substituted benzene ring); 1H NMR (DMSO-d6, 400 MHz): δH (ppm) 0.98 (d, J = 6.5 Hz, 3H, CH3), 3.34 (s, 1H, N–CH), 3.38 (m, 2H, N–CH2), 3.46 (t, J = 6.7 Hz, 2H, CO–CH2), 5.19 (bs, 1H, OH), 6.24 (d, 1H, J = 3.8 Hz, O–CH), 7.30–8.67 (m, 9H, aromatic) and 8.78 (bs, 2H, NH2+); 13C-NMR (DMSO-d6, 400 MHz): δC (ppm) 9.82 (CH3), 35.43 (N–CH), 39.99 (CO–CH2), 58.57 (N–CH2), 70.05 (O–CH), 122.73, 126.27, 127.87, 128.21, 128.65, 131.22, 134.60, 137.61, 141.30, 148.46 (aromatic carbons) and 195.98 (CO); MS-ESI: m/z at 329 [M + H]+; anal. calcd for C18H21ClN2O4: C, 59.26; H, 5.80; N, 7.68. Found C, 59.40; H, 5.82; N, 7.71.
N-((1S,2S)-1-Hydroxy-1-phenylpropan-2-yl)-3-(4-methoxyphenyl)-3-oxopropan-1-amine HCl (3g). White crystalline powder; yield 86%; mp: 156–158 °C; [α]25D: +40.4° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC); IR (KBr, cm−1): purity: 99+% (HPLC); ee 100% (chiral HPLC, RT 14.24 min); IR (KBr, cm−1): 3298 (b, OH), 3032 and 2912 (NH2+), 1688 (CO), 1589 and 1491 (benzenoid), 1312 (C–N), 1213 (C–O), 783 (di substituted benzene ring), and 702 (mono substituted benzene ring); 1H-NMR (DMSO-d6, 400 MHz): δH (ppm) 0.95 (d, J = 6.7 Hz, 3H, CH3), 3.38 (t, J = 6.8 Hz, 2H, CO–CH2), 3.52 (m, 1H, N–CH), 3.55 (t, J = 6.9 Hz, 2H, N–CH2), 3.84 (s, 3H, OCH3), 5.21 (bs, 1H, OH), 6.16 (d, J = 4.1 Hz, 1H, O–CH), 7.06–7.97 (m, 9H, aromatic protons) and 8.92 (bs, 2H, NH2+); 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 9.78 (CH3), 34.72 (CO–CH), 40.46 (N–CH), 56.08 (N–CH2), 58.65 (OCH3), 69.95 (O–CH), 114.52, 126.29, 127.76, 128.61, 129.39, 130.80, 141.57, 163.95 (aromatic carbons) and 195.86 (CO); MS-ESI: m/z at 314.1 [M + H]+; anal. calcd for C19H24ClNO3: C, 65.52; H, 6.91; N, 4.00. Found C, 65.59; H, 6.96; N, 4.02.
N-((1R,2R)-1-Hydroxy-1-phenylpropan-2-yl)-3-oxo-3-phenylpropan-1-amine HCl (3a′). White crystalline powder; yield 90%; mp: 111–113 °C; [α]25D: −56.4° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 7.67 min); IR (KBr, cm−1): 3331 (broad, OH), 2818 and 2767 (NH2+), 1688 (CO), 1584 and 1450 (benzenoid), 1342 (C–N), 1215 (C–O), 739, 706 and 689; 1H-NMR (DMSO-d6, 400 MHz): δH (ppm) 0.96 (d, J = 6.7 Hz, 3H, CH3), 3.39 (m, 1H, N–CH), 3.41 (t, J = 2.4 Hz, 2H, CO–CH2), 3.61 (t, J = 6.9 Hz, 2H, N–CH2), 5.23 (bs, 1H, OH), 6.16 (d, J = 2.8 Hz, 1H, O–CH), 7.32–7.97 (m, 10H, aromatic protons), 9.04 (bs, 2H, NH2+); 13C-NMR (DMSO-d6, 400 MHz): δC (ppm) 9.73 (CH3), 35.16 (CO–CH2), 39.82 (N–CH), 58.67 (N–CH2), 69.88 (O–CH), 126.27, 127.71, 128.39, 128.58, 129.32, 134.13, 136.41, 141.61 (aromatic carbons), 197.47 (CO); MS-ESI: m/z at 284 [M + H]+; anal. calcd for C18H22ClNO2: C, 67.60; H, 6.93; N, 4.38. Found C, 67.68; H, 7.00; N, 4.40.
N-((1R,2R)-1-Hydroxy-1-phenylpropan-2-yl)-3-oxo-3-p-tolylpropan-1-amine HCl (3b′). White crystalline powder; yield 90%; mp: 205–206 °C; [α]25D: −49.2° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 9.29 min); IR (KBr, cm−1): 3319 (broad, OH), 2980 and 2830 (NH2+), 1688 (CO), 1607 and 1541 (benzenoid), 1381 (C–N), 1221 (C–O), 787 (p-disubstituted benzene ring), and 704 (mono substituted benzene ring); 1H NMR (DMSO-d6, 400 MHz): δH (ppm) 0.94 (d, J = 6.3 Hz, 3H, CH3), 2.37 (s, 3H, Ph–CH3), 3.38 (t, J = 4 Hz, 2H, CO–CH2), 3.42 (dd, J = 5.7 & 2.9 Hz, 1H, N–CH), 3.50 (t, J = 6.5 Hz, 2H, N–CH2), 5.17 (bs, 1H, OH), 6.18 (d, J = 3.5 Hz, 1H, O–CH), 7.27–7.89 (m, 9H, aromatic protons) and 8.71 (bs, 2H, NH2+), 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 9.94 (CH3), 21.65 (Ph–CH3), 34.97 (CO–CH2), 40.40 (N–CH), 58.63 (N–CH2), 70.06 (O–CH), 126.30, 127.90, 128.56, 128.68, 129.89, 133.94, 141.35, 144.75 (aromatic carbons) and 197.05 (CO); MS-ESI: m/z at 298 [M + H]+; anal. calcd for C19H24ClNO2: C, 68.35; H, 7.25; N, 4.20. Found C, 68.43; H, 7.28; N, 4.22.
3-(4-Chlorophenyl)-N-((1R,2R)-1-hydroxy-1-phenyl-propan-2-yl)-3-oxopropan-1-amine HCl (3c′). White crystalline powder; yield 87%; mp: 142–143 °C; [α]25D: −37.4° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 9.30 min); IR (KBr, cm−1): 3227 (b, OH), 2978, and 2850 (aliphatic and aromatic), 2737 and 2677 (NH2+), 1674 (CO), 1585 and 1537 (benzenoid), 1338 (C–N), 1221 (C–O), 982, 760 (o,p-di substituted benzene ring) and 696 (mono substituted benzene ring); 1H-NMR (DMSO-d6, 400 MHz): δH (ppm) 1.03 (d, J = 6.7 Hz, 3H, CH3), 3.41 (t, J = 15.7 & 8.8 Hz, 1H, CO–CH), 3.47 (t, J = 6.4 Hz, 2H, N–CH), 3.68 (t, J = 7.0 Hz, 2H, N–CH2), 5.31 (bs, 1H, OH), 6.24 (d, J = 4.1 Hz, 1H, O–CH), 7.33–8.07 (m, 9H, aromatic protons) and 9.12 (bs, 2H, NH2+); 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 9.79 (CH3), 35.28 (CO–CH2), 40.20 (N–CH), 58.72 (CO–CH2), 69.96 (O–CH), 126.35, 127.80, 128.66, 129.51, 130.41, 135.21, 139.06, 141.67 (aromatic carbons) and 196.56 (CO); MS-ESI: m/z 318 & 320 in the ratio of 3:1 respectively; anal. calcd for C18H21Cl2NO2: C, 61.02; H, 5.97; N, 3.95. Found C, 61.26; H, 6.00; N, 3.99.
3-(2,4-Dichlorophenyl)-N-((1R,2R)-1-hydroxy-1-phenylpropan-2-yl)-3-oxopropan-1-amine HCl (3d′). White crystalline powder; yield 89%; mp: 196–197 °C; [α]25D: −25.2° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 8.40 min); IR (KBr, cm−1): 3337 (b, OH), 2826 and 2785 (NH2+), 1699 (CO), 1584 and 1551 (benzenoid), 1313 (C–N), 1206 (C–O), 980 and 824 (tri substituted benzene ring), and 708 (mono substituted benzene ring); 1H NMR (DMSO-d6, 400 MHz): δH (ppm) 1.01 (d, J = 6.6 Hz, 3H, CH3); 3.59 (t, 2H, CO–CH2), 3.62 (dd, J = 8.1 & 4.1 Hz, 1H, N–CH), 3.66 (m, 2H, N–CH2), 5.31 (bs, 1H, OH), 6.26 (d, J = 4.1 Hz, 1H, O–CH), 7.31–7.90 (m, 8H, aromatic protons) and 9.20 (bs, 2H, NH2+); 13C NMR (DMSO-d6, 400 MHz): δC (ppm) 9.73 (CH3), 52.31 (CO–CH2), 58.77 (N–CH), 69.88 (N–CH2), 71.71 (O–CH), 126.28, 127.74, 128.22, 128.61, 130.71, 131.20, 131.78, 136.40, 137.23, 141.62 (aromatic carbons) and 198.30 (CO); MS-ESI: m/z at 352, 354 & 356 [M + H]+ in the ratio of 9:6:1 respectively; anal. calcd for C18H20Cl3NO2: C, 55.62; H, 5.19; N, 3.60. Found C, 55.80; H, 5.22; N, 3.62.
N-((1R,2R)-1-Hydroxy-1-phenylpropan-2-yl)-3-(3-hydroxyphenyl)-3-oxopropan-1-amine HCl (3e′). White crystalline powder; yield 94%; mp: 174–176 °C; [α]25D: −58.7° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 6.24 min); IR (KBr, cm−1): 3344 (b, OH), 2799 and 2738 (NH2+), 1681 (CO), 1584 and 1498 (benzenoid), 1316 (C–N), 1232 (C–O), 855, 770 (tri substituted benzene ring) and 703 (mono substituted benzene ring); 1H NMR (DMSO-d6, 400 MHz): δH (ppm) 0.97 (d, J = 6.5 Hz, 3H, CH3), 3.41 (t, J = 6.2 Hz, 2H, CO–CH2), 3.56 (s, 2H, N–CH), 3.70 (m, 1H, N–CH2), 5.21 (bs, 1H, OH), 6.19 (s, 1H, O–CH), 7.09–7.45 (m, 9H, aromatic protons), 8.84 (2H, NH2+) and 10.03 (bs, 1H, Ph–OH); 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 9.86 (CH3), 35.20 (CO–CH2), 58.62 (N–CH2), 69.99 (O–CH), 114.62, 119.33, 121.25, 126.31, 127.81, 128.64, 130.43, 137.80, 141.55, 158.19 (aromatic carbons) and 197.35 (CO); MS-ESI: m/z at 300 [M + H]+; anal. calcd for C18H22ClNO3: C, 64.38; H, 6.60; N, 4.17. Found C, 64.55; H, 6.63; N, 4.20.
N-((1R,2R)-1-Hydroxy-1-phenylpropan-2-yl)-3-(3-nitrophenyl)-3-oxopropan-1-amine HCl (3f′). White crystalline powder; yield 90%; mp: 164–165 °C; [α]25D: −57.4° (c = 1%, methanol); purity: 99+% (HPLC); ee > 99.9% (chiral HPLC, RT 8.71 min); IR (KBr, cm−1): 3373 (b, OH), 2975 and 2761 (NH2+), 1696 (CO), 1584 and 1495 (benzenoid), 1528 and 1351 (NO2), 1314 (C–N), 1202 (C–O), 767 (disubstituted benzene ring) and 704 (mono substituted benzene ring); 1H NMR (DMSO-d6, 400 MHz): δH (ppm) 1.00 (dd, J = 30.4 & 6.7 Hz, 3H, CH3), 3.26 (m, 1H, N–CH), 3.42 (t, J = 8 Hz, 2H, CO–CH2), 3.72 (t, J = 6.8 Hz, 2H, N–CH2), 4.50 (bs, 1H, OH), 6.49 (d, J = 89.7 Hz, 1H, O–CH), 7.38–8.69 (m, 9H, aromatic protons) and 9.27 (bs, 2H, NH2+); 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 9.82 (CH3), 35.43 (CO–CH2), 39.99 (N–CH2), 58.57 (N–CH2), 70.05 (O–CH), 122.73, 126.27, 127.87, 128.21, 128.65, 131.22, 134.60, 137.61, 141.30, 148.46 (aromatic carbons) and 195.98 (CO); MS-ESI: m/z at 329 [M + H]+; anal. calcd for C18H21ClN2O4: C, 59.26; H, 5.80; N, 7.68. Found C, 59.42; H, 5.82; N, 7.69.
N-((1R,2R)-1-Hydroxy-1-phenylpropan-2-yl)-3-(4-methoxyphenyl)-3-oxopropan-1-amine HCl (3g′). White crystalline powder; yield 87%; mp: 155–157 °C; [α]25D: −39.2° (c = 1%, methanol); purity: 99+% (HPLC); ee 100% (chiral HPLC, RT 16.22 min); IR (KBr, cm−1): 3298 (b, OH), 3059 and 2916 (NH2+), 1688 (CO), 1587 and 1448 (benzenoid), 1310 (C–N), 1213 (C–O), 771 (disubstituted benzene ring) and 702 (mono substituted benzene ring); 1H-NMR (DMSO-d6, 400 MHz): δH (ppm) 0.98 (d, J = 6.7 Hz, 3H, CH3); 3.41 (t, J = 3.5 Hz, 2H, CO–CH2), 3.56 (m, 1H, N–CH), 3.58 (t, J = 7.3 Hz, 2H, N–CH2), 3.86 (s, 3H, OCH3), 5.24 (bs, 1H, OH), 6.17 (d, 1H, O–CH), 7.09–8.00 (m, 9H, aromatic protons) and 8.92 (bs, 2H, NH2+); 13C-NMR (DMSO-d6, 100 MHz): δC (ppm) 9.82 (CH3), 34.77 (CO–CH2), 40.49 (N–CH), 56.10 (N–CH2), 58.67 (OCH3), 69.99 (O–CH), 114.54, 126.31, 127.77, 128.62, 129.41, 130.81, 141.61, 163.97 (aromatic carbons) and 195.89 (CO); MS-ESI: m/z at 314 [M + H]+; anal. calcd for C19H24ClNO3: C, 65.23; H, 6.91; N, 4.00. Found C, 65.46; H, 6.95; N, 4.03.
Anti-proliferative analysis
HepG-2, A549 and Vero cells were purchased from ATCC, Manassas, VA, USA. Anti-proliferative properties of synthesised compounds 3a–g and 3a′–g′ were studied against HepG-2 and A549 cells. HepG-2 and A549 cells were maintained in DMEM and Vero cells maintained in RPMI with 10% fetal bovine serum and 2 mM L-glutamine, along with antibiotics. The anti-proliferative activity was determined according to established method.44 Aqueous one solution reagent was used according to the manufacturer's guidelines. The percentage of growth inhibition was calculated using the following formula: inhibition (%) = A − B/A × 100 (A – control group and B – treated group).
Immunofluorescence studies
Treated and untreated HepG-2 cells were washed twice with ice-cold PBS and suspended in binding buffer for 20 min. Cells were incubated with FITC, PI and DAPI for 30 min at 4 °C in the dark condition. Cells were then centrifuged and pellets were smeared. FITC, PI and DAPI fluorescences were immediately observed under confocal microscope (Zeiss, 2009).
Colony formation assay
HepG-2 cells (500 cells per well) were seeded in 12 well tissue culture plates overnight. Treated and untreated cells were incubated for 10 days at 37 °C in 5% CO2. Colonies were then washed twice in PBS and stained with crystal violet for 1 h and photos were taken.
Apoptosis studies
Annexin V-FITC/PI detection kit was used for apoptosis analysis using flow cytometry as per manufacturer's guidelines. Briefly, cells were harvested after treatment and resuspended in binding buffer. Aliquots of 105 cells were mixed with 5 μL of annexin V-FITC and 15 μL of PI solutions for 15 min at room temperature in the dark. After incubation, 400 μL binding buffer was added, and cells were analyzed by FACS Calibur flow cytometer (Becton Dickinson).
Cell cycle analysis
Cell cycle analysis was performed using a CycleTEST™ PLUS DNA reagent kit according to the manufacturer's protocol. Briefly, cells were centrifuged for 5 min at 300 × g at room temperature. The supernatant was aspirated leaving approximately 50 μL of residual fluid in the tube to avoid disturbing the pellet. 1 mL of buffer solution was added and the cells were resuspended by gently vortexing at low speed. Cells were centrifuged for 5 min at 300 × g at room temperature. 250 μL of solution A was added to cell pellets and kept for 10 min at room temperature. 200 μL of solution B was added and kept for 10 min at room temperature. Finally solution C was added and kept for 10 min in dark on ice and immediately the samples were run on the flow cytometer (Becton Dickinson).
Western blot
Western blot analysis was performed following the method of Balachandran et al., (2015).45
In vivo model
Animal experiments were approved by Regulations for the Management of Laboratory Animals at Fujita Health University, Japan. The procedure for the ethical use of these animals was approved by the Animal Care and Use Committee at Fujita Health University (approval no: M3291). Fundamental guidelines for proper conduct of animal experiments and related activities in academic research institutions under the Jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology. Four to five weeks old BALB/c SIc-nu/nu female mice were purchased form Chubu kagaku Shizai Co., Ltd (Nagoya city, Japan). HepG-2 cells were harvested from sub-confluent cultures and washed with serum free medium. Only suspensions consisting of single cells, with >90% viability, were used for the injections. 1 × 107 cells (100 μL, media:Matrigel, 1:1) were injected subcutaneously into the right flank. After a week of implantation, tumor diameters were measured (Mitutoyo Company, Kawasaki, Japan). When tumors reached 100 mm3 in diameter, the mice were randomized into two groups based on the tumor volume. Group-I was treated with PBS (100 μL; i.p.; 3 times per week), group II was treated with 3a (30 mg kg−1; i.p.; 3 times per week). Treatment was continued for up to three weeks from the date of randomization (day 0). The tumor volume was measured at three day intervals. The mice were killed 22 days after randomization. The tumors were carefully excised and the tumor volume was measured. Samples were prepared for western blot analysis. The xenograft tumor volume was calculated as V = 1/2 × (length × width2).
Western blot analysis for tumor tissues
TRIZOL reagent was used for protein extraction from HepG-2 tumor tissues as per manufacturer's guidelines. Isolated proteins were subjected to western blot analysis.
Immunohistochemistry analysis
The tumor samples were embedded in paraffin, cut into 5 μm sections and stained with hematoxylin and eosin (H & E). All comparisons of staining intensities were made at 200× magnifications.
Docking analysis
Molecular docking analysis was evaluated as described previously.45 Briefly, the protein structures of Bcl-2 (PDB ID: 4IEH) and JAK-2 (PDB ID: 2XA4) were downloaded from the Protein Data Bank. AutoDock version 4.2.5.1 and AutoDock Tools (ADT) version 1.5.6 were used for docking studies. Initially, the crystal water was removed using ADT, the polar hydrogen atoms were added, Gasteiger charges were added to the each atom and merged with non-polar hydrogen atoms. The distance between donor and acceptor atoms that formed a hydrogen bond was defined as 1.9 Å with a tolerance of 0.5 Å, and the acceptor–hydrogen–donor angle was not less than 120°. All the structures were saved in PDBQT file format for further studies in ADT. A grid box with dimension of 40 × 40 × 40 Å3 and 50 × 40 × 40 Å3, with 0.375 Å spacing and centered on (x, y, z) 0.488/4.140/25.437 and 12.153/25.794/11.850 that included the active site of JAK-2 and Bcl-2, respectively. Then, the calculation of grid energy was carried out. Docking study was analyzed by an empirical-free energy function and genetic algorithm. An each ligand was examined twenty independent docking studies and receptor-ligand adduct for lowest free energy of binding conformation from the largest cluster and saved in PDBQT format. In this study, genetic algorithms were used for energy calculations. Finally, the outputs of binding modes, polar and hydrophobic interactions were expressed using PyMol software.
Author contributions
C. B. and K. C. R. contributed equally to this work.
Conflict of interest
The authors state no conflict of interest.
Abbreviations
| PPA | Phenylpropanolamine |
| Ψ-PPA | Pseudophenylpropanolamine |
| STAT | Signal transducer and activator of transcription |
| JAK | Janus kinase |
| HepG-2 | Hepatocellular carcinoma |
| A549 | Lung cancer |
| DMEM | Dulbecco's modified eagle medium |
| PBS | Phosphate buffer saline |
| FITC | Fluorescein isothiocyanate |
| PI | Propidium iodide |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| FBS | Fetal bovine serum |
| ATCC | American type culture collection |
| SDS-PAGE | Sodium dodecyl sulphate polyacrylamide gel electrophoresis |
| TLC | Thin layer chromatography |
| HPLC | High-performance liquid chromatography |
| UV | Ultraviolet |
| NMR | Nuclear magnetic resonance |
| FT-IR | Fourier transform infrared |
| ADT | AutoDock tools |
| SOR | Specific optical rotations |
Acknowledgements
The authors sincerely thank Fujita Health University, Toyoake, Aichi, Japan for research support and financial assistance. The authors also thank the management of Malladi Drugs & Pharmaceuticals Ltd. Chennai, India for providing support to conduct this research activity. We thank Dr Y. Yamamoto for providing JAK/STAT antibody for protein expression studies. We thank Ms Sachiko Iba and Ms Keiko Hattori for helping in confocal, western blot and animal studies.
References
- N. A. Flavahan, J. Pharmacol. Exp. Ther., 2005, 313, 432–439 CrossRef CAS PubMed.
- O. K. Haugeto, K. E. Schroder and I. W. Mair, J. Otolaryngol., 1981, 10, 359–362 CAS.
- K. Toll and P. Graf, Rhinology, 2006, 44, 274–277 Search PubMed.
- J. Maryadele O'Neil, The Merck Index: An Encyclopedia of Chemicals, Drugs & Biological, Merck Research Laboratories, NJ, USA, 14th edn, 2006 Search PubMed.
- I. Declerck, B. Himpens, G. Droogmans and R. Casteels, Pfluegers Arch., 1990, 417, 117–119 CrossRef CAS PubMed.
- M. M. Weinberger, Pediatr. Clin. North Am., 1975, 22, 121–127 CrossRef CAS PubMed.
- H. I. Silverman, B. E. Kreger, G. P. Lewis, A. Karabelas, R. Paone and M. Foley, Curr. Ther. Res., 1984, 28, 185–194 Search PubMed.
- J. Loman, M. Rinkel and A. Myerson, Am. Heart J., 1939, 18, 89–93 CrossRef.
- C. R. Lake, S. Gallant, E. Masson and P. Miller, Am. J. Med., 1990, 89, 195–208 CrossRef CAS PubMed.
- J. S. Walker, J. Pharm. Sci., 1989, 78, 986–989 CrossRef CAS PubMed.
- P. R. Pentel, R. W. Asinger and N. L. Benowitz, Clin. Pharmacol. Ther., 1985, 37, 488–494 CrossRef CAS PubMed.
- M. J. Ellenhorn and D. G. Barceloux, Medical Toxicology – Diagnosis and Treatment of Humna Poisoning, Elsevier Science Publishing Co. Inc, New York, NY, 1988, p. 293 Search PubMed.
- K. Mazumdar, N. K. Dutta, K. A. Kumar and S. G. Dastidar, Biol. Pharm. Bull., 2005, 28, 713–717 CAS.
- W. Osswald and S. Guimaraes, Naunyn-Schmiedeberg's Arch. Pharmacol., 1971, 270, 203–209 CrossRef CAS PubMed.
- J. Engel, K. Axel, K. Posselt, F. Stroman and K. Thiemer, Cycloalphatic ketomaines, US Pat., US4542159, 1985.
- K. Thiele, US Pat., US3225095A, 1965.
- C. L. Wainwright and J. R. Parratt, Eur. J. Pharmacol., 1988, 147, 373–380 CrossRef CAS PubMed.
- S. K. Wan, W. Hoff and T. R. Evans, Curr. Med. Res. Opin., 1988, 11, 242–253 CrossRef CAS PubMed.
- H. C. Caldwell, US Pat., US3769317, 1973.
- S. Iwamato and W. H. Hartung, J. Org. Chem., 1994, 9, 513–517 CrossRef.
- K. Chennakesava Rao, Y. Arun, K. Easwaramoorthi, C. Balachandran, T. Praksam, T. E. Yuvaraj and P. T. Perumal, Bioorg. Med. Chem. Lett., 2014, 34, 3057–3063 CrossRef PubMed.
- K. Chennakesava Rao, Y. Arun, K. Easwaramoorthi, C. Balachandran, K. S. Muralithara Rao, M. Govindan, E. Nobuhiko, T. Prakasm and P. T. Perumal, Bioorg. Med. Chem. Lett., 2015, 25, 4232–4238 CrossRef CAS PubMed.
- N. Samie, S. Muniandy, M. S. Kanthimathi, B. S. Haerian and R. E. Azudin, Sci. Rep., 2016, 6, 29056 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- Cancer I. A. f. R. o. & Organization, W. H. GLOBOCAN: Estimated Cancer Incidence, Mortality, and Prevalence Worldwide in 2012, IARC, 2014 Search PubMed.
- M. Benekli, M. R. Baer, H. Baumann and M. Wetzler, Blood, 2003, 101, 2940–2954 CrossRef CAS PubMed.
- E. Darnell Jr, I. M. Kerr and G. R. Stark, Science, 1994, 264, 1415–1421 Search PubMed.
- J. F. Bromberg, M. H. Wrzeszczynska, G. Devgan, Y. Zhao, R. G. Pestell, C. Albanese and J. E. Darnell Jr, Cell, 1999, 98, 295–303 CrossRef CAS PubMed.
- H. Yu, D. Pardoll and R. Jove, Nat. Rev. Cancer, 2009, 9, 798–809 CrossRef CAS PubMed.
- J. Lee, J. C. Kim, S. E. Lee, C. Quinley, H. Kim, S. Herdman, M. Corr and E. Raz, J. Biol. Chem., 2012, 287, 18182–18189 CrossRef CAS PubMed.
-
(a) E. Schmidt, Arch. Pharm., 1906, 244, 239–240 CrossRef CAS;
(b) H. Emde and E. Schmidt, Arch. Pharm., 1906, 244, 269–299 CrossRef.
-
(a) E. Hermann, Helv. Chim. Acta, 1929, 12, 365–376 CrossRef;
(b) E. Hermann, Helv. Chim. Acta, 1929, 12, 377–384 CrossRef.
- T. Jerphagnon, A. J. A. Gayet, F. Berthiol, V. Ritleng, N. Mrsiu, A. Meetsma, M. Pfeffer, A. J. Minnaard, B. L. Feringa and J. G. de Vries, Chem.–Eur. J., 2009, 15, 12780–12790 CrossRef CAS PubMed.
- K. Thiele, K. Posselt, H. Offermanns and K. Theimer, Arzneimittel Forschung, 1980, 30, 747–751 CAS.
-
(a) K. Thiele, US Pat., US3621052, 1971;
(b) K. Thiele, US Pat., US3646145, 1972.
-
(a) K. Posselt, B. Enkhelm and K. Thiele, US Pat., US3658845, 1972;
(b) K. Thiele, E. Koberstein and G. Nonnenmacher, Arzneimittel Forschung, 1968, 18, 1255–1263 CAS.
- J. Engel, A. Kleemann, K. Posselt, F. Stroman and K. Thiemer, US Pat., US4542159, 1985.
- G. M. Rusch, G. M. Hoffman, R. F. McConnell and W. E. Rinehart, Toxicol. Appl. Pharmacol., 1986, 83, 69–78 CrossRef CAS PubMed.
- T. R. Torkelson, S. E. Sadek and V. K. Rowe, Am. Ind. Hyg. Assoc. J., 1961, 22, 263–270 CrossRef PubMed.
- ESI†.
- C. Balachandran, N. Emi, Y. Arun, Y. Yamamoto, B. Ahilan, B. Sangeetha, V. Duraipandiyan, Y. Inaguma, A. Okamoto, S. Ignacimuthu, N. A. Al-Dhabi and P. T. Perumal, Chem.-Biol. Interact., 2015, 242, 81–90 CrossRef CAS PubMed.
- P. Sansone and J. J. Bromberg, Clin. Oncol., 2012, 30, 1005–1014 CrossRef CAS PubMed.
- K. Sakamoto, B. A. Creamer, A. A. Triplett and K. Wagner, Mol. Endocrinol., 2007, 21, 1877–1892 CrossRef CAS PubMed.
- J. J. Westendorf, G. J. Ahmann, P. R. Greipp, T. E. Witzig, J. A. Lust and D. F. Jelinek, Leukemia, 1996, 10, 866–876 CAS.
- C. Balachandran, N. Emi, Y. Arun, N. Yamamoto, V. Duraipandiyan, Y. Inaguma, A. Okamoto, S. Ignacimuthu, N. A. Al-Dhabi and P. T. Perumal, Chem.-Biol. Interact., 2016, 249, 23–35 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1443803. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra22480f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 c,
N. Emia,
N. Yamamotod,
Y. Inagumaa,
A. Okamotoa,
K. Easwaramoorthib and
P. T. Perumalc
c,
N. Emia,
N. Yamamotod,
Y. Inagumaa,
A. Okamotoa,
K. Easwaramoorthib and
P. T. Perumalc