
Cytotoxicity studies of coumarin analogs: design, synthesis and biological activity
Abstract
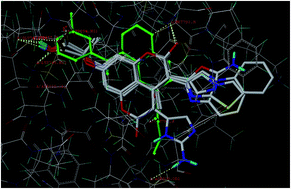
In the present study, a series of coumarin derivatives were designed, synthesized and evaluated for their antioxidant and cytotoxic properties. The title compounds, 2-(3-substituted-4-methyl-2-oxo-2H-chromen-7-yloxy)-2-methylpropanoic acid derivatives 5a–5f, were synthesized by base-catalyzed dehydrohalogenative cyclization following Hantzsch synthesis. All the newly synthesized analogues were characterized and established on the basis of mass, 1H NMR, 13C NMR and IR studies. The compounds were evaluated for their in vitro antioxidant activity and found to exhibit substantial activity. The in vitro cytotoxicity was evaluated against MCF-7, MDA-231 (human breast cancer) and HT29 (human colon adenocarcinoma) cell lines by MTT assay and the results were encouraging. Compound 5b, with lower IC50 values of 2.4 and 4.8 μM for MCF-7 and MDA-231, respectively, was considered to be potent among the series.
 Please wait while we load your content...
Please wait while we load your content...

