DOI:
10.1039/C6RA22462H
(Paper)
RSC Adv., 2016,
6, 92259-92266
In situ formation of MoS2/C nanocomposite as an anode for high-performance lithium-ion batteries†
Received
8th September 2016
, Accepted 20th September 2016
First published on 20th September 2016
Abstract
Anode materials with excellent electrochemical properties as an alternative to carbon-based structures are suggested for advanced high-performance lithium-ion batteries. Here, composites containing MoS2 and carbon (MoS2/C) were in situ synthesized via heat treatment at 700 °C under a CH4 atmosphere with varying reaction times. XRD, Raman, XPS, and TEM data show that the MoS2/C composites consist of crystalline MoS2 and an amorphous carbon phase and show a homogeneous distribution of curved and bent MoS2 particles with a carbon matrix. In particular, the MoS2/C composite with an optimal content of the amorphous carbon phase exhibits relatively an excellent performance in lithium-ion batteries, facilitating the lithiation/delithiation process in MoS2 as an electroactive material.
1. Introduction
Lithium-ion batteries (LIBs) have been spotlighted as main electrochemical power sources, which require both high energy and high power density. For the electrochemical power sources, novel electrode materials and structures are required and should be proposed.1–3 However, since graphite as an anode exhibits a relatively low capacity of 372 mA h g−1, anode materials having a high reversible energy density have been suggested.4,5 Therefore, alternative anode materials with high specific energy density and improved cycle properties need to be proposed for LIBs.6 Among the promising candidates, MoS2 has recently exhibited high reversible capacity and improved cycling performance. This metal sulfide as MoS2, which is analogous to a graphene structure, consists of S–Mo–S layers linked by van der Waals forces.7,8 It has been reported that the MoS2 structure could be responsible for the improved Li intercalation/deintercalation without a significant volumetric variation.9,10 However, nevertheless, MoS2-based electrodes exhibit poor electrical conductivity and the existence of polysulfides Li2Sx during the cycling.11–15
Recently, it was reported that nanostructure electrodes of MoS2 with conductive materials such as carbon could exhibit improved structural and electrochemical properties in LIBs.16–24 In general, the incorporation of conducting agents such as various carbon materials with MoS2 (MoS2/carbon) can lead to a remarkably improved cycling performance and high-rate capability, including nanosheet, nanosphere, and nanotube structure. MoS2/carbon nanocomposite electrodes have exhibited improved LIB performance in comparison with bulk-type MoS2 anodes.25–35 Furthermore, the capacity of the MoS2/graphene nanocomposite can reach 1483 mA h g−1, exhibiting an improved capacity retention of 91.1%.36 Gong et al. prepared 3D structures from 2D MoS2-nanosheets, which showed an enhanced cyclability of 1220 mA h g−1 at 74 mA g−1 within 30 cycles.37 Herein, we reported the in situ formation of MoS2/C composites as anodes for use in high-performance LIBs via heating process at 700 °C in a CH4 atmosphere with varying reaction times. The structural and chemical analysis of MoS2/C composites was performed using field-emission scanning electron microscopy (FE-SEM), field-emission transmission electron microscopy (FE-TEM), X-ray diffraction (XRD), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS). The charge/discharge curves, cycling performance, and Nyquist plots of the as-prepared anode materials were obtained using lithium coin cells.
2. Experimental
2.1 Synthesis of MoS2 and MoS2/C
For the preparation of MoS2, 0.88 g of ammonium molybdate ((NH4)6Mo7O2·4H2O, Sigma-Aldrich) and 2.64 g of sodium sulfide nonahydrate (Na2S·9H2O, Sigma-Aldrich) were mixed in 0.8 M HCl solution at 80 °C with continuous stirring. 0.7 g of hydroxylamine hydrochloride (NH2OH·HCl, Sigma-Aldrich) was added to the mixed solution at 80 °C for 1 h. After the complete reaction, the dark brown precipitate as a precursor for MoS2 was washed with de-ionized water and ethanol and dried in 60 °C oven. Finally, to obtain the nanocomposite, the resulting powder was heated at 700 °C with varying reaction times of 1, 3, and 5 h under CH4 atmosphere (denoted as MoS2/C-1, MoS2/C-3, and MoS2/C-5, respectively).38 For comparison, the resulting powder was heated at 500 °C for 2 h under N2 atmosphere (denoted as MoS2-only).
2.2 Structural analysis
The size and morphology of the samples were characterized using a field-emission scanning electron microscope (FE-SEM, JSM-7800F, JEOL Ltd) and a field-emission transmission electron microscope (FE-TEM, Tecnai G2 F30 system). The crystal structure was observed using an X-ray diffractometer (XRD, D2 PHASER, Bruker AXS, Cu Kα (λ = 0.15418 nm) source, a with a nickel filter). The scan was carried out in the 2θ range from 10° to 80° with a scan rate of 0.5° min−1. Thermogravimetric analysis (TGA) curves were observed using a thermal analyzer (SDTA851, Mettler Toledo) in the range of 25 to 700 °C at a heating rate of 5 °C min−1 under an air flow of 60 cm3 min−1. Raman spectra were recorded on a high resolution Mirco-Raman spectrometer (LabRam Aramis, Horriba Jovin Yvon) using an Ar ion laser with 532.8 nm. X-ray photoelectron spectroscopy analysis (XPS, Thermo VG, U.K.) was carried out using an Al Kα X-ray source of 1486.6 eV at a chamber pressure below 10−8 Torr and a beam power of 200 W. The electrical resistance of the pellets under compression was measured in the current–voltage plots using potentiostat at room temperature. Nitrogen adsorption and desorption isotherms were measured using a Micromeritics ASAP 2020. The samples were outgassed at 573 K for 6 h.
2.3 Electrochemical analysis
To prepare lithium coin cells (size 2032, Hohsen Co.), the MoS2/C composite samples as a working electrode were assembled with lithium metal foil (FMC Co.) as a counter electrode. To prepare the electrode materials, the active material (80 wt%), ketjen black (10 wt%), and a binder (10 wt%, polyvinylidene difluoride (PVDF), Alfa Aesar) were mixed in 1-methyl-2-pyrrolidinone solvent (99%, Aldrich). The paste was deposited onto a copper metal foil and then dried in a 100 °C vacuum oven. The loading amount of anode materials for all electrodes was 0.6–0.7 mg cm−2. Furthermore, to assemble coin-type full cell with LiCoO2 cathode and MoS2 anode, the slurries for cathode and anode consisted of 90 and 80 wt% active materials, 5 and 10 wt% PVDF, and 5 and 10 wt% ketjen black, respectively. The cells were assembled inside an argon-filled glove box (<5 ppm, H2O and O2). The polypropylene membrane (Celgard 2400) was inserted between electrodes. The electrolyte solution consists of 1.1 M LiPF6 in a solvent mixture of ethylene carbonate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dimethyl carbonate = 1:1 (Soulbrain Co., Ltd). The charge–discharge measurement was carried out using a multichannel battery tester (WBCS300L, Wonatech Co.). The cycle performance of the half cells was measured between 0.01 and 3.0 V vs. Li/Li+ with varying currents with a rest period of 30 min at room temperature. The interface resistance and Li diffusivity of the cells were observed using electrochemical impedance spectroscopy (EIS).
:dimethyl carbonate = 1:1 (Soulbrain Co., Ltd). The charge–discharge measurement was carried out using a multichannel battery tester (WBCS300L, Wonatech Co.). The cycle performance of the half cells was measured between 0.01 and 3.0 V vs. Li/Li+ with varying currents with a rest period of 30 min at room temperature. The interface resistance and Li diffusivity of the cells were observed using electrochemical impedance spectroscopy (EIS).
3. Results and discussion
Fig. 1 shows the XRD patterns of MoS2-only, MoS2/C-1, MoS2/C-3, and MoS2/C-5. XRD peaks at 14.8°, 32.6°, 39.5°, 49.7°, and 58.3° corresponding to (002), (100), (103), (105), and (110), respectively, of the hexagonal MoS2 phase (JCPDS #37-1492) with lattice parameters of a = 3.16 Å, b = 3.16 Å, and c = 12.30 Å without diffraction peaks related to impurities or molybdenum oxides. In addition, the diffraction peaks related to carbon could not be observed due to an amorphous carbon phase formed under CH4 atmosphere at a relatively low temperature of 700 °C. This shows that all as-prepared samples form a well-formed molybdenum disulfide with amorphous carbon.39 The electrodes having amorphous carbon materials can exhibit an improved cycling performance due to alleviated volume expansions of the electrodes.40 In the XRD patterns, it is noticeable that the XRD intensity of the (002) planes for the MoS2/C composite electrodes is weaker than that of MoS2-only having the strongest (002) plane intensity. In general, it was reported that a well-stacked molybdenum disulfide structure into a c-axis exhibited a strong intensity of (002).41,42 Hu et al. also observed that the gradual peak narrowing of the (002) in the XRD patterns was attributed to the decreased number of (002) planes in carbon coated MoS2, leading to ultrathin MoS2 nanostructures.43 Accordingly, the relatively low intensity of (002) in MoS2/C implies that the MoS2/C composites have less stacking layers of (002) and thus might consist of smaller sized MoS2 particles than those with MoS2-only.
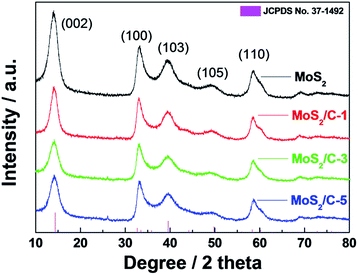 |
| | Fig. 1 XRD patterns of the as-prepared samples. | |
Raman spectra of the as-prepared samples were obtained, as indicated in Fig. 2. The characteristic peaks of MoS2 and carbon were observed. The E12g at 383 cm−1 corresponds to the vibration of two S atoms with respect to the Mo atom. The A1g at 408 cm−1 is associated with the out-of-plane vibration of only S atoms in opposite directions, were detected (Fig. 2(a)).44,45 Furthermore, the MoS2/C-1, MoS2/C-3, and MoS2/C-5 showed two characteristic carbon peaks containing the G and D band at ∼1592 and ∼1356 cm−1, respectively (Fig. 2(b)). In the Raman spectra, the G band is associated with the E12g vibration mode of the sp2 carbon crystal structures, whereas the D band reflects the structural defects on the graphitic plane.46–48 The relative intensity ratio (ID/IG) values, of MoS2/C-1, MoS2/C-3, and MoS2/C-5 are 0.98, 1.11 and 1.14, respectively. From XRD and Raman analysis, it is concluded that the composites consist of crystalline MoS2 and amorphous carbon phases.
 |
| | Fig. 2 Raman spectra of the as-prepared samples. | |
To elucidate the chemical states of the as-prepared samples, XPS peaks were observed as indicated in Fig. S1.† Fig. S1(a)–(d)† show the spectra of Mo 3d in the samples. The two main characteristic peaks corresponding to the Mo 3d5/2 and 3d3/2 of Mo4+ in MoS2 are located at ∼229.5 and ∼232.6 eV, respectively. Furthermore, the Mo 3d peaks contain small portions of Mo 3d5/2 and 3d3/2 of Mo6+ in MoO3. The S 2s peak is located at ∼226.5 eV. The binding energies of the S 2p peaks in the samples consist of S 2p3/2 and 2p1/2 of S2−, which are located at ∼162.5 and ∼163.6 eV, respectively (Fig. S1(e)–(h)).† The C 1s XPS spectrum of the MoS2-only exhibits the main peak at ∼284.6 eV related to the C–C bonds, while the C 1s spectra of the composites contains the C–S peaks at ∼286.3 eV as well as the main peaks at ∼284.6 eV related to the C–C bonds (Fig. S1(i)–(l)†). As shown in Fig. S2† of the mapping images in the as-prepared samples, the MoS2-only exhibited a homogeneous distribution of sulphur and molybdenum. On the other hand, the MoS2/C-1, MoS2/C-3, and MoS2/C-5 displayed a good dispersion of sulphur, molybdenum, and carbon within the composite structures.
As shown in Fig. 3 of FE-TEM images, the MoS2-only seemed to have an irregular size and straight sheet shape with a layered structure. The high-resolution TEM (HR-TEM) images dominantly showed over 40 layers and fine lattice fringes of 0.62 nm corresponding to the (002) plane of the MoS2 phase. In contrast, the as-prepared composites exhibited the particles with curved and folded layers, which result from internal stress caused by the composite structure with carbon. In the HR-TEM images, the MoS2 phases consisted of 10–15 layers corresponding to the (002) planes with d-spacing of 0.62 nm, while the carbon showed an amorphous structure on the MoS2 surface. Fig. S3† shows nitrogen adsorption/desorption isotherms of the samples. The specific surface areas of MoS2-only, MoS2/C-1, MoS2/C-3, and MoS2/C-5 were 118.0, 101.8, 93.3, and 88.4 m2 g−1, respectively. With increasing reaction time for synthesis of composite, MoS2/C composites exhibited decreased specific surface area and narrow pore size distribution, as compared to MoS2-only, represents the formation of dense composite structure. The formation of a carbon phase during the heating process at 700 °C under a CH4 atmosphere prevents the aggregation of the MoS2 particles, thus resulting in a homogeneous carbon structure and size distribution. Accordingly, the in situ formation of the following the MoS2/C nanostructure may occur according to eqn (1):49
| | |
MoS2 + CH4 → MoS2 + C + 2H2 → MoS2/C
| (1) |
 |
| | Fig. 3 TEM images of MoS2-only ((a) and (b)), MoS2/C-1 ((c) and (d)), MoS2/C-3 ((e) and (f)), and MoS2/C-5 ((g) and (h)). | |
The amount of carbon in the composites was determined using TGA as shown in Fig. S4.† The initial weight reduction in the range of 25 to 200 °C is associated with the evaporation of H2O and solvent. The second weight reduction at around 280 to 400 °C is related to the oxidation of MoS2 to MoO3 and the subsequent weight gain after 400 °C is due to the combustion of carbon (C + O2 → CO2).50 The carbon contents in MoS2/C-1, MoS2/C-3, and MoS2/C-5 were determined to be 2.8, 7.5, and 14.0 wt%, respectively. As shown in Fig. S5,† the MoS2-only appears to have a relatively low electrical conductivity (2.37 × 10−4 S cm−1) whereas the MoS2/C composites show an improved conductivity (0.13–5.5 S cm−1), which may be due to the existence of a carbon phase as a conductor, compared to the MoS2-only having semiconductor properties.
Therefore, as indicated in Fig. 4, the MoS2/C composites consisting of MoS2 and carbon could be in situ prepared via facile synthesis at 700 °C under a CH4 atmosphere with varying reaction times. In particular, the composite structures having a crystalline electroactive material as a core (MoS2) and an amorphous carbon phase as an electronic conductor can be favorable due to the small-sized electroactive materials and thin stacking layers used to facilitate lithium ion transport with the shorter diffusion channels. Furthermore, the composite with an appropriate portion of carbon phase, especially, the MoS2/C-3, can provide an effective conductive network between the MoS2 particles and improve the transporting of the electrolyte in the electrodes, thus showing an enhanced LIB performance during the cycling process.43
 |
| | Fig. 4 An illustration of the in situ formation of MoS2/C via heat treatment at 700 °C under a CH4 atmosphere with different reaction times. | |
As shown in Fig. 5, all the 1st discharge profiles contain two plateaus at ∼1.2 and ∼0.7 V, suggesting the formation of LixMoS2 during the two-step conversion reaction process. In the following cycles, two plateaus at ∼2.0 and ∼1.2 V are observed. However, the charge curves of all the samples exhibited one plateau at ∼2.3 V associated with a reversible lithium-ion behavior. Typically, the theoretical capacity of MoS2 as a main active material can be calculated as follows:51
| | |
MoS2 + xLi+ + xe− → LixMoS2 (1.1 V vs. Li/Li+) (167 mA h g−1)
| (2) |
| | |
LixMoS2 + (4 − x)Li+ + (4 − x)e− → Mo + 2Li2S (0.6 V vs. Li/Li+) (669 mA h g−1)
| (3) |
| | |
S + 2Li+ + 2e− → Li2S (2.2 V vs. Li/Li+) (1675 mA h g−1)
| (4) |
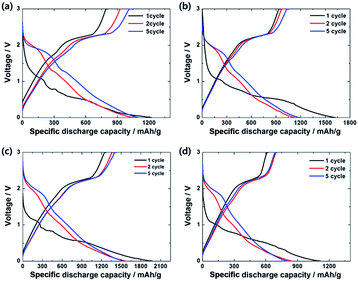 |
| | Fig. 5 Charge–discharge curves of (a) MoS2-only, (b) MoS2/C-1, (c) MoS2/C-3, and (d) MoS2/C-5 during the cycling between 3.0 and 0 V at 200 mA g−1. | |
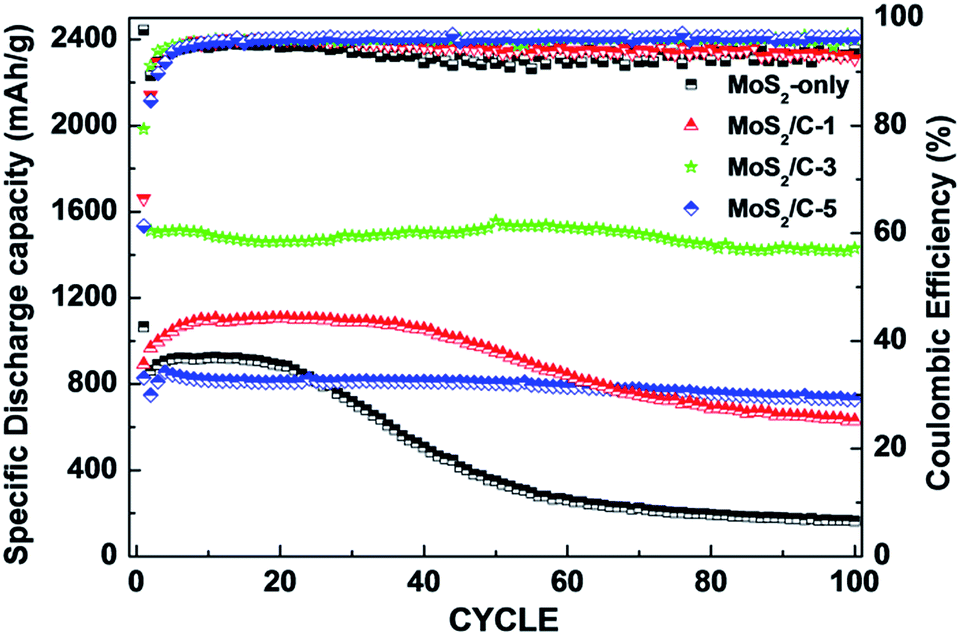
The 1st discharge capacities of MoS2-only, MoS2/C-1, MoS2/C-3, and MoS2/C-5 were 1225.9, 1623.9, 1978.1, and 1119.2 mA h g−1, respectively, which were equivalent to that of theoretical MoS2 structure as an anode.51 In contrast, MoS2-only, MoS2/C-1, MoS2/C-3, and MoS2/C-5 exhibited 100th discharge capacities of 323.1, 436.4, 1742.7, and 721.7 mA h g−1, respectively, and the capacity retention ratios (100th/2nd cycle) of 30.7%, 38.6%, 110.7%, and 85%, respectively. Among the samples, the MoS2/C-3 composite showed the highest discharge capacities and improved retention ratio during the cycle process.
As indicated in Fig. 6, the MoS2/C composites exhibited high discharge capacities and improved retention ratios after 100 cycles in comparison with MoS2-only. Furthermore, the coulombic efficiencies of MoS2-only, MoS2/C-1, MoS2/C-3, and MoS2/C-5 after 100 cycles were 94%, 92%, 96%, and 96%, respectively. The morphology of the samples was observed before and after the cycling at 500 mA g−1 (Fig. S6†). The MoS2-only seemed to be cracked after the cycling process whereas MoS2/C electrodes maintained relatively stable morphology. It is likely that the sudden capacity reduction of MoS2-only is ascribed to the lack of electronic conducting network and pulverization caused by the volume expansion during the cycling process. It is significant that the MoS2/C composite anodes with increased carbon contents revealed highly enhanced cycle life and specific capacity retention. Especially, among the composites, the MoS2/C-3 with an appropriate amount of amorphous carbon matrix as a conductive network displayed better electrochemical properties during the cycling process, which is mainly attributed to enhanced electronic and ionic conduction. The current rates were elevated from 100 to 1000 mA g−1 (Fig. 7(a)) and from 2 to 20 A g−1 (Fig. 7(b)). Among these samples, the MoS2/C-3 composite showed the highest specific capacity at each current rate, i.e. improved relative retention of the discharge capacity. In particular, the specific discharge capacities of MoS2/C-3 at 2, 10, and 20 A g−1 are 948.7, 672.8, and 436.7 mA h g−1, respectively, representing an improved rate cyclability in LIBs.
 |
| | Fig. 6 Plots of specific discharge capacity and coulombic efficiency vs. cycle number of the samples for 100 cycles at a current rate of 500 mA g−1. | |
 |
| | Fig. 7 Rate cycling performance of the samples from (a) 100 to 1000 mA g−1 and from (b) 2 to 20 A g−1. | |
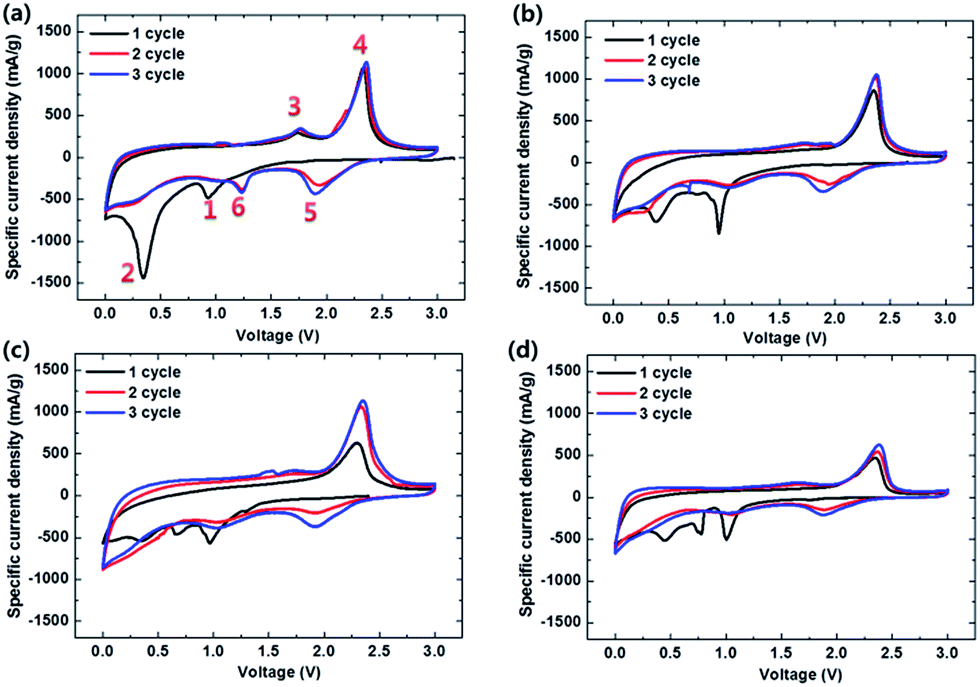
In the 1st CV curves of Fig. 8, the electrodes show a reduction peak at 0.9 V: (1) MoS2 + xLi+ + xe− → LixMoS2. The characteristic peak at 0.3 V represented the following reaction: (2) LixMoS2 + (4 − x)Li+ + (4 − x)e− → Mo + 2Li2S. The 1st and 2nd anodic peaks at 1.7 and 2.3 V showed (3) the slight partial oxidation of Mo and (4) Li2S− + 2e− → 2Li+ + S, respectively. In the 2nd cycles, two reversible reduction peaks at ∼2.0 and ∼1.2 V are assigned to the reactions of (5) S + 2Li+ + 2e− → Li2S and (6) MoS2 + xLi+ + xe− → LixMoS2, respectively. However, the reduction peaks of (1) and (2) disappeared from the 2nd cycles of all the electrodes. The CVs of the samples remained steady after the 1st cycle, exhibiting both excellent reversibility and stability during the insertion/extraction process.
 |
| | Fig. 8 CVs of (a) MoS2-only, (b) MoS2/C-1, (c) MoS2/C-3, and (d) MoS2/C-5 measured after 1st, 2nd, and 3rd cycles in the voltage range of 3.0 and 0 V vs. Li/Li+ at a scan rate of 0.2 mV s−1. | |
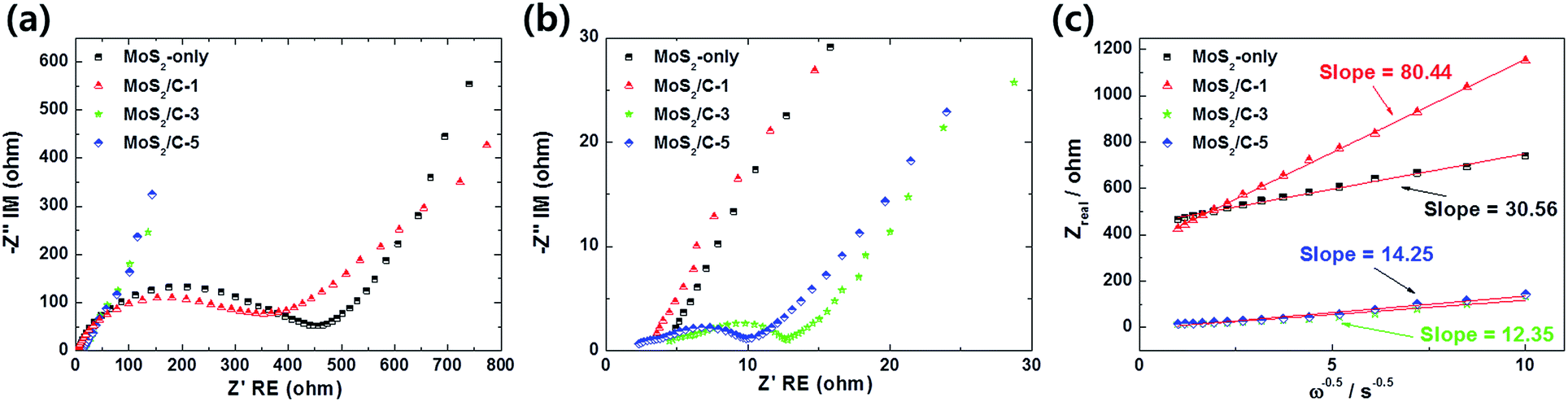
Nyquist plots of the anodes were obtained after the 5th cycle at 2.5 V, as shown in Fig. 9. In the plots, the semicircles at the high and medium frequencies are ascribed to the resistance of the solid-electrolyte-interface film and charge transfer resistance (Rct) between the electrode-electrolyte interface, respectively. In addition, the straight lines in the low frequency are ascribed to the lithium-ion movement in the anodes. After the 5th cycle, the values of Rct of MoS2-only, MoS2/C-1, MoS2/C-3, and MoS2/C-5 were 412, 359, 7.6, and 7 Ω, respectively. The MoS2/C composite anodes exhibited lower Rct due to an improved electronic conduction by the carbon matrix formed on MoS2 compared to MoS2-only. The plots of ZRe vs. square root of frequency (ω−1/2) in the low frequency range are shown in Fig. 9(c). Using the relationship between ZRe and ω−1/2, the lithium-ion diffusion coefficient (DLi) can be determined using the following eqn (5):
| |
 | (5) |
here,
R is the gas constant (8.314 J K
−1 mol
−1),
T is 298 K,
F is the Faraday constant (96
485 C mol
−1),
A is the surface area of the electrode (1.3273 cm
2),
n is the number of electrons involved in reaction,
C is the concentration of lithium ion, and
σ is the Warburg coefficient, which can be obtained from the line of
ZRe ∼
ω−1/2. The
C could be calculated from the density and the molecular weight of the material.
52 The value of
C is 0.00769 mol cm
−3 in this work. The
DLi for MoS
2-only, MoS
2/C-1, MoS
2/C-3, and MoS
2/C-5 is determined as 3.64 × 10
−13, 5.26 × 10
−14, 2.23 × 10
−12, and 1.68 × 10
−12 cm
2 s
−1, respectively. The incorporation of carbon in the composites can provide the improved conductivity of electrons and the fast transport of lithium-ions during the cycle process. In particular, MoS
2/C-3 exhibited the lowest resistance and highest Li-ion diffusion coefficient, resulting in significantly improved electrochemical properties. Thus, the enhanced cell performance of the MoS
2/C composites as anodes for lithium-ion reaction result from the reduced transfer resistance and high lithium-ion diffusivity in the composite structure consisting of MoS
2 as an active material and carbon as an electrical conductor. In particular, MoS
2/C-3 exhibited excellent LIB performance during the lithiation/delithiation process due to the MoS
2 structure with curved layers and an optimal amount of carbon as a conductive matrix. Also, it was reported that MoS
2-based anodes fabricated using various carbon structures such as graphene and carbon cloth exhibited improved electrochemical performance.
53,54
 |
| | Fig. 9 Nyquist plots of the samples obtained after 5th cycle at 2.5 V (a) between 100 kHz and 10 mHz and (b) in the lower frequency range. (c) Plots of ZRe vs. square root of frequency (ω−1/2) for the samples in the low frequency range. | |
Typically, the anodes in LIBs consists of electroactive materials (80 wt%, MoS2-only and MoS2/C-3), a binder (10 wt%), and a conducting agent (10 wt%). Here, we fabricated conducting agent-free electrode structures (f-MoS2/C-3) fabricated using MoS2/C-3 with PVDF as a binder in the absence of a conductive material as illustrated in Fig. 10(a). For comparison, the electrode was prepared in the presence of a conductive material (c-MoS2/C-3), as indicated in Fig. 10(b). The thicknesses of the electrodes were ∼6 (f-MoS2/C-3) and ∼15 μm (c-MoS2/C-3), respectively, as observed in the SEM images. The relatively thinner f-MoS2/C-3 means that the conducting agent free-electrode structure can have a higher packing density based on the same amount of an active material in the electrodes, compared to c-MoS2/C-3 containing 10 wt% carbon black, producing highly improved volumetric properties.
 |
| | Fig. 10 (a) Conducting agent-free electrode structure without a conducting agent (f-MoS2/C-3) and (b) conventional electrode structure with a conducting agent (c-MoS2/C-3) and plots of (c), (d) mass and (e) volumetric discharge capacity vs. cycle number for f-MoS2/C-3, f-MoS2-only, and c-MoS2/C-3 at 500 mA g−1 for 50 cycles. | |
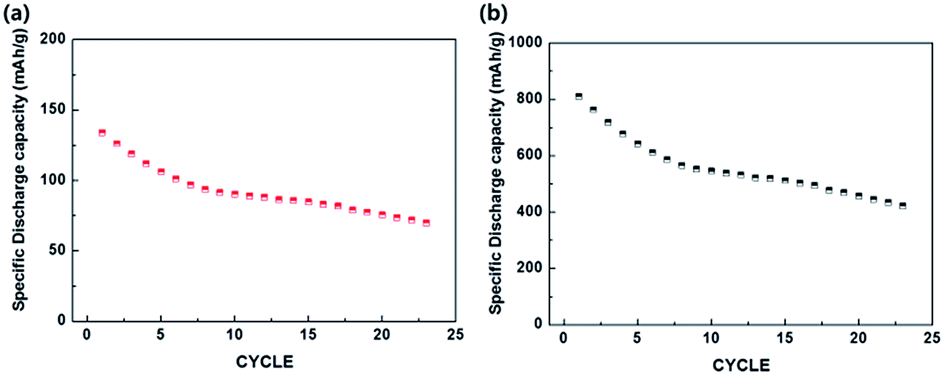
The f-MoS2/C-3 showed superior cycling properties at 500 mA g−1 for 50 cycles compared to f-MoS2-only (Fig. 10(c)). In the conducting agent-free electrodes, the improved performance of f-MoS2/C-3 results from the existence carbon phase as a conductor. Furthermore, as shown in Fig. 10(d) and (e), f-MoS2/C-3 exhibited reversible specific mass capacity of 307 mA h g−1 and volumetric capacity of 741 mA h cm−3. In particular, the high volumetric capacity of f-MoS2/C-3 can leads to an efficient utilization of active materials within a given volume in LIBs. For the LIB test of full cell (denoted as LiCoO2/MoS2/C-3), LiCoO2 and MoS2/C-3 were utilized as cathode and anode, respectively. As shown in Fig. 11, in LiCoO2/MoS2/C-3, the 1st discharge capacity based on cathode weight is ∼134 mA h g−1 whereas the 1st discharge capacity based on anode weight is ∼810 mA h g−1. Typically, it was reported that commercial-type full cell of LiCoO2/Graphite exhibited a discharge capacity of ∼130 mA h g−1.55 However, an optimized electrode structure and fabrication procedure will remain as further works.
 |
| | Fig. 11 Plots of (a) cathode based and (b) anode based specific discharge capacity vs. cycle number of the full cell at a current rate of 0.15 C-rate. | |
4. Conclusions
In summary, we reported in situ formation MoS2/C composites as anodes for use in high-performance LIBs via heat treatment at 700 °C under a CH4 atmosphere with varying reaction times. The composites consisted of crystalline MoS2 and amorphous carbon phase and showed the homogeneous mixture of curved and bent MoS2 particles in a carbon matrix. The MoS2/C composites exhibited relatively high capacity, improved capacity retention, and high rate cycling compared with MoS2-only. Specifically, the excellent LIB performance of MoS2/C-3, containing an optimal portion of carbon as an electronic conductive material, can be ascribed to the facilitation of the curved MoS2 structure for lithiation and the synergistic effect between the MoS2 with carbon.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A2012541).
Notes and references
- K. Kang, Y. S. Meng, J. Bréger, C. P. Grey and G. Ceder, Science, 2006, 311, 977–980 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695–1715 CrossRef CAS PubMed.
- H. S. S. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., 2010, 49, 4059–4062 CrossRef PubMed.
- A. Li, H. Liu, Z. Zhu, M. Huang and Y. Yang, J. Mater. Sci. Technol., 2006, 22, 40–44 CAS.
- H. Hwang, H. Kim and J. Cho, Nano Lett., 2011, 11, 4826–4830 CrossRef CAS PubMed.
- S. Ding, D. Zhang, J. S. Chen and X. W. Lou, Nanoscale, 2012, 4, 95–98 RSC.
- W. Ahn, K.-B. Kim, K.-N. Jung, K.-H. Shin and C.-S. Jin, J. Power Sources, 2012, 202, 394–399 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- D. Wang, A. Fu, H. Li, Y. Wang, P. Guo, J. Liu and X. S. Zhao, J. Power Sources, 2015, 285, 469–477 CrossRef CAS.
- Q. Liu, Z. Wu, Z. Ma, S. Dou, J. Wu, L. Tao, X. Wang, C. Ouyang, A. Shen and S. Wang, Electrochim. Acta, 2015, 177, 298–303 CrossRef CAS.
- J.-M. Jeong, K. G. Lee, S.-J. Chang, J. W. Kim, Y.-K. Han, S. J. Lee and B. G. Choi, Nanoscale, 2015, 7, 324–329 RSC.
- L. Zhang and X. W. Lou, Chem.–Eur. J., 2014, 20, 5219–5223 CrossRef CAS PubMed.
- M. Gu, Y. Li, X. Li, S. Hu, X. Zhang, W. Xu, S. Thevuthasan, D. R. Baer, J. G. Zhang, J. Liu and C. Wang, ACS Nano, 2012, 6, 8439–8447 CrossRef CAS PubMed.
- E. J. Yoo, J. Kim, E. Hosono, H. S. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed.
- D. N. Futaba, K. Hata, T. Yamada, T. Hiraoka, Y. Hayamizu, Y. Kakudate, O. Tanaike, H. Hatori, M. Yumura and S. Iijima, Nat. Mater., 2006, 5, 987–994 CrossRef CAS PubMed.
- H. Li, Z. Wang, L. Chen and X. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- M. Yoshio, H. Wang, K. Fukuda, Y. Hara and Y. Adachi, J. Electrochem. Soc., 2000, 147, 1245 CrossRef CAS.
- S. L. Candelaria, Y. Shao, W. Zhou, X. Li, J. Xiao, J.-G. Zhang, Y. Wang, J. Liu, J. Li and G. Cao, Nano Energy, 2012, 1, 195–220 CrossRef CAS.
- L. Zhang, W. Fan, W. W. Tjiu and T. Liu, RSC Adv., 2015, 5, 34777–34787 RSC.
- L. Zhang, W. Fan and T. Liu, RSC Adv., 2015, 5, 43130–43140 RSC.
- S. K. Das, R. Mallavajula, N. Jayaprakash and L. A. Archer, J. Mater. Chem., 2012, 22, 12988 RSC.
- H. Jiang, D. Ren, H. Wang, Y. Hu, S. Guo, H. Yuan, P. Hu, L. Zhang and C. Li, Adv. Mater., 2015, 27, 3687–3695 CrossRef CAS PubMed.
- B. Guo, K. Yu, H. Fu, Q. Hua, R. Qi, H. Li, H. Song, S. Guo and Z. Zhu, J. Mater. Chem. A, 2015, 3, 6392–6401 CAS.
- X. Li, W. Li, M. Li, P. Cui, D. Chen, T. Gengenbach, L. Chu, H. Liu and G. Song, J. Mater. Chem. A, 2015, 3, 2762–2769 CAS.
- X. Wang, Z. Zhang, Y. Chen, Y. Qu, Y. Lai and J. Li, J. Alloys Compd., 2014, 600, 84–90 CrossRef CAS.
- G. Li, X. Zeng, T. Zhang, W. Ma, W. Li and M. Wang, CrystEngComm, 2014, 16, 10754–10759 RSC.
- N. Lingappan, N. H. Van, S. Lee and D. J. Kang, J. Power Sources, 2015, 280, 39–46 CrossRef CAS.
- J. Li, Y. Hou, X. Gao, D. Guan, Y. Xie, J. Chen and C. Yuan, Nano Energy, 2015, 16, 10–18 CrossRef CAS.
- R. Wang, C. Xu, J. Sun, Y. Liu, L. Gao, H. Yao and C. Lin, Nano Energy, 2014, 8, 183–195 CrossRef CAS.
- X. Zhou, L.-J. Wan and Y.-G. Guo, Nanoscale, 2012, 4, 5868–5871 RSC.
- X. Xu, Z. Fan, X. Yu, S. Ding, D. Yu and X. W. Lou, Adv. Energy Mater., 2014, 4, 1–5 Search PubMed.
- Y. Liu, Y. Zhao, L. Jiao and J. Chen, J. Mater. Chem. A, 2014, 2, 13109 CAS.
- Y. Gong, S. Yang, L. Zhan, L. Ma, R. Vajtai and P. M. Ajayan, Adv. Funct. Mater., 2014, 24, 125–130 CrossRef CAS.
- H. Yang, S. Liu, J. Li, M. Li, G. Peng and G. Zou, Nanotechnology, 2006, 17, 1512–1519 CrossRef CAS.
- H. Liu, F. Zhang, W. Li, X. Zhang, C.-S. Lee, W. Wang and Y. Tang, Electrochim. Acta, 2015, 167, 132–138 CrossRef CAS.
- J. Yu, J. Yang, X. Feng, H. Jia, J. Wang and W. Lu, Ind. Eng. Chem. Res., 2014, 53, 12697–12704 CrossRef CAS.
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720–4728 CrossRef CAS PubMed.
- G. Du, Z. Guo, S. Wang, R. Zeng, Z. Chen and H. Liu, Chem. Commun., 2010, 46, 1106–1108 RSC.
- S. Hu, W. Chen, J. Zhou, F. Yin, E. Uchaker, Q. Zhang and G. Cao, J. Mater. Chem. A, 2014, 2, 7862 CAS.
- H. Li, Q. Zhang, C. C. R. Yap, B. K. Tay, T. H. T. Edwin, A. Olivier and D. Baillargeat, Adv. Funct. Mater., 2012, 22, 1385–1390 CrossRef CAS.
- Q. Li, E. C. Walter, W. E. Van Der Veer, B. J. Murray, J. T. Newberg, E. W. Bohannan, J. A. Switzer, J. C. Hemminger and R. M. Penner, J. Phys. Chem. B, 2005, 109, 3169–3182 CrossRef CAS PubMed.
- J. Schwan, S. Ulrich, V. Batori, H. Ehrhardt and S. Silva, J. Appl. Phys., 1996, 80, 440 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, ACS Nano, 2014, 8, 5069–5078 CrossRef CAS PubMed.
- A. Ferrari, J. C. Meyer, C. Scardaci, C. Casiraghi and M. Lazzeri, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- A. Venugopal, S. Naveen Kumar, J. Ashok, D. Hari Prasad, V. Durga Kumari, K. B. S. Prasad and M. Subrahmanyam, Int. J. Hydrogen Energy, 2007, 32, 1782–1788 CrossRef CAS.
- X. Xiong, W. Luo, X. Hu, C. Chen, L. Qie, D. Hou and Y. Huang, Sci. Rep., 2015, 5, 9254 CrossRef CAS PubMed.
- T. Stephenson, Z. Li, B. Olsen and D. Mitlin, Energy Environ. Sci., 2014, 7, 209 CAS.
- L. Li, Z. Chen, Q. Zhang, M. Xu, X. Zhou, H. Zhu and K. Zhang, J. Mater. Chem. A, 2015, 3, 894–904 CAS.
- H. Yu, C. Zhu, K. Zhang, Y. Chen, C. Li, P. Gao, P. Yang and Q. Ouyang, J. Mater. Chem. A, 2014, 2, 4551–4557 CAS.
- H. Yu, C. Ma, B. Ge, Y. Chen, Z. Xu, C. Zhu, C. Li, Q. Ouyang, P. Gao, J. Li, C. Sun, L. Qi, Y. Wang and F. Li, Chem.–Eur. J., 2013, 19, 5818–5823 CrossRef CAS PubMed.
- Z. Hu, Q. Liu, W. Sun, W. Li, Z. Tao, S. Chou, J. Chen and S. X. Dou, Inorg. Chem. Front., 2016, 3, 532–535 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra22462h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.