
New cadinane sesquiterpenoids from the basidiomycetous fungus Pholiota sp.†
Jie
Lin
a,
Renlei
Wang
a,
Guohua
Xu
a,
Zhengfeng
Ding
a,
Xueshen
Zhu
a,
Xingzhong
Liu
b,
Jian
Zou
c,
Guodong
Chen
c,
Li
Li
d and
Ling
Liu
*b
aJiangsu Key Laboratory for Biofunctional Molecules, College of Life Science and Chemistry, Jiangsu Second Normal University, 77 West Beijing Road, Nanjing 210013, P. R. China
bState Key Laboratory of Mycology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, P. R. China. E-mail: liul@im.ac.cn.; Tel: +86 10 64806153
cInstitute of Traditional Chinese Medicine & Natural Products, College of Pharmacy, Jinan University, Guangzhou 510632, P. R. China
dInstitute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100050, P. R. China
First published on 24th November 2016
Abstract
Five new cadinane sesquiterpenoids, pholiotins A–E (1–5), and three known compounds, 11-hydroxy-1(10)-valencen-2-one (6), 8,11-dihydroxy-1(10)-eremophilen-2-one (7) and durgamone (8), were isolated from the crude extract of Pholiota sp. Their structures were established on the basis of extensive spectroscopic analysis. Their absolute configurations were determined by X-ray diffraction, the Snatzke's method and electronic circular dichroism (ECD) calculations. All isolates were tested for antifungal activity.
Introduction
Fungi are well-known producers of bioactive secondary metabolites.1–5 Based on this consideration and the documented success in finding new bioactive natural products from special types of fungi,6 we initiated chemical studies of the Cordyceps-colonizing fungi,7–10 and those that were isolated from the soil samples surrounding Cordyceps sinensis.11,12 In the current work, the basidiomycetous fungus Pholiota sp. (SCK05-7-ZP19) was isolated from a soil sample on the surface of C. sinensis collected in Kangding, Sichuan, People's Republic of China. Some species of Pholiota are edible, but some are toxic.13–16 The isolated fungus Pholiota sp. (SCK05-7-ZP19) was grown in a solid-substrate fermentation culture. An organic solvent extract of the culture was active against Aspergillus flavus (CGMCC 3.0951). Bioassay-guided fractionation of the extract led to the isolation of five new metabolites, pholiotins A–E (1–5), and three known compounds, 11-hydroxy-1(10)-valencen-2-one (6),17 8,11-dihydroxy-1(10)-eremophilen-2-one (7),18 and durgamone (8)19,20 (Fig. 1). Details of the isolation, structure elucidation, and antifungal activity of these metabolites are reported herein. | ||
| Fig. 1 Chemical structure of 1–8. | ||
Results and discussion
Pholiotin A (1) was assigned the molecular formula C15H24O5 (four degrees of unsaturation) on the basis of HRESIMS. The 1H and 13C NMR spectra (Table 1) of 1 showed resonances for two methyl groups, four methylenes (one oxygenated), five methines (one oxygenated), two olefinic carbons (one protonated), one quaternary carbon (δC 74.9) and one carboxylic carbon (δC 168.7). These data accounted for all 1H and 13C resonances except for four exchangeable protons, and suggested that 1 was a bicyclic compound. Interpretation of the 1H–1H COSY NMR data established two isolated proton spin-systems (Fig. 2), which were C-3–C-2–C-1–C-6–C-7–C-8–C-9 and C-7–C-12 (including C-13 and C-14). HMBC correlations (Fig. 2) from H-1, and H2-8 to C-10, and from H2-15 to C-1, C-9, and C-10 implied that C-1, C-9 and C-15 were all attached to C-10 directly, completing the cyclohexane moiety. Correlations from H2-2 to C-4, H2-3 to C-5, and from H-5 to C-1, C-3 and C-7 revealed the connections of the C-4/C-5 olefin to C-3, and C-6, respectively, establishing the cyclohexene ring, which fused to the cyclohexane subunit at C-1/C-6 to the completion of the octahydronaphthalene core structure. Other correlations from H-3b and H-5 to the carboxylic carbon C-11 (δC 168.7) showed that C-11 was connected to the C-4/C-5 olefin at C-4. Considering the chemical shifts for C-9 (δC 69.5), C-10 (δC 74.9), C-11 (δC 168.7) and C-15 (δC 62.8), as well as the molecular formula requirement for 1, the remaining four exchangeable protons were assigned as C-9-, C-10-, C-11- and C-15-OH, respectively. Therefore, the gross structure of pholiotin A was established as 1. Finally, the structure of 1 was confirmed by single-crystal X-ray crystallographic analysis, and a perspective ORTEP plot is shown in Fig. 3. The X-ray data also allowed assignment of its relative configuration.| Pos. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δ C a | δ H b (J in Hz) | δ C a | δ H b (J in Hz) | δ C a | δ H b (J in Hz) | |
| a Recorded at 125 MHz. b Recorded at 500 MHz. | ||||||
| 1 | 42.1, CH | 1.80, m | 43.0, CH | 1.80, m | 43.0, CH | 1.80, m |
| 2a | 22.4, CH2 | 2.24, m | 22.2, CH2 | 2.25, m | 22.3, CH2 | 2.24, m |
| 2b | 1.28, m | 1.28, m | 1.28, m | |||
| 3a | 26.4, CH2 | 2.47, m | 26.3, CH2 | 2.47, m | 26.4, CH2 | 2.50, m |
| 3b | 2.18, m | 2.18, m | 2.18, m | |||
| 4 | 131.7, qC | 131.9, qC | 131.7, qC | |||
| 5 | 140.6, CH | 7.07, s | 140.3, CH | 7.10, s | 140.4, CH | 7.11, s |
| 6 | 40.2, CH | 2.04, m | 40.4, CH | 2.04, m | 40.4, CH | 2.04, m |
| 7 | 39.5, CH | 1.66, m | 40.1, CH | 1.66, m | 40.1, CH | 1.66, m |
| 8a | 28.4, CH2 | 1.83, m | 26.6, CH2 | 1.81, m | 28.5, CH2 | 1.81, m |
| 8b | 1.52, m | 1.52, m | 1.52, m | |||
| 9 | 69.5, CH | 4.07, br s | 73.1, CH | 5.37, br s | 69.5, CH | 4.02, br s |
| 10 | 74.9, qC | 74.2, qC | 73.7, qC | |||
| 11 | 168.7, qC | 168.5, qC | 168.5, qC | |||
| 12 | 26.5, CH | 2.12, m | 26.5, CH | 2.12, m | 26.5, CH | 2.12, m |
| 13 | 21.5, CH3 | 0.92, d (6.9) | 21.5, CH3 | 0.95, d (6.9) | 21.5, CH3 | 0.97, d (6.9) |
| 14 | 15.4, CH3 | 0.82, d (6.9) | 15.2, CH3 | 0.87, d (6.9) | 15.4, CH3 | 0.88, d (6.9) |
| 15a | 62.8, CH2 | 3.63, br s | 63.1, CH2 | 3.71, d (12) | 62.8, CH2 | 4.22, d (12) |
| 15b | 3.65, d (12) | 4.16, d (12) | ||||
| 16 | 171.0, qC | |||||
| 17 | 20.8, CH3 | 2.15, s | ||||
| 18 | 170.9, qC | |||||
| 19 | 21.2, CH3 | 2.12, s | ||||
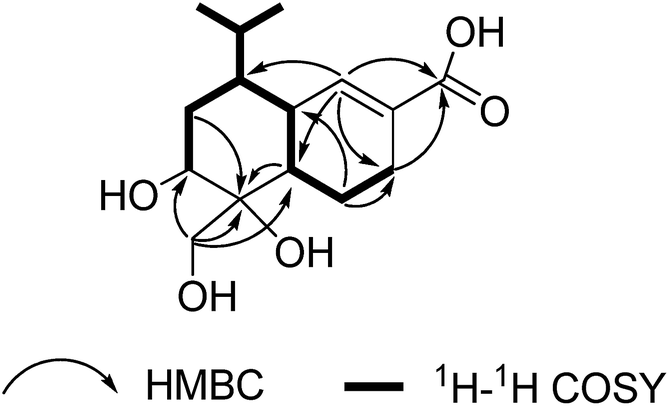 | ||
| Fig. 2 Key 1H–1H COSY, HMBC correlations for 1. | ||
 | ||
| Fig. 3 Thermal ellipsoid representations of 1 and 5. | ||
The absolute configuration of 1 was initially assigned by application of the modified Mosher method.21,22 Treatment of 1 with (S)- and (R)-MTPACl afforded the (R)-MTPA ester 1a and the (S)-MTPA esters 1b, respectively. The selectivity for the acylation of C-9 hydroxy group was not achieved, but the ester 1a acylation of C-15 hydroxy group was obtained by reversed-phase HPLC purification. The absolute configuration of the sec/tert-9,10-diol moiety in 1a was assigned using the in situ dimolybdenum CD method developed by Frelek.23,24 Upon addition of dimolybdenum tetraacetate [Mo2(OAc)4] to 1a in DMSO solution, a metal complex of chiral vic-diol with the achiral Mo2(OAc)4 was generated as an auxiliary chromophore. Since 1a has an inherent CD resulting from the C-11 carboxylic chromophore, this contribution was subtracted to give the induced CD of the metal complex to avoid its overlap (>250 nm) with those generated after addition of Mo2(OAc)4. Therefore, the observed sign of the Cotton effect in the induced spectrum originates solely from the chirality of the vic-diol moiety expressed by the sign of the O–C–C–O torsion angle. The positive Cotton effects observed at around 310 and 400 nm, respectively, in the induced CD spectrum permitted assignment of the 9S and 10S absolute configuration in 1a on the basis of the empirical rule proposed by Snatzke (Fig. 4 and 5). The absolute configuration of 9,10-diol moiety in 1 was deduced as 9S and 10S by analogy to 1a. Considering the relative configuration established by single-crystal X-ray crystallographic, 1 was assigned the 1S,6S,7R,9S, and 10S.
 | ||
| Fig. 4 CD spectrum of 1a in DMSO containing Mo2(OAc)4. | ||
 | ||
| Fig. 5 The sterical configurations for the cottonogenic derivative of 1a. | ||
The absolute configuration of 1 was further confirmed by comparison of the experimental and the simulated circular dichroism (CD) spectra (Fig. 6) generated by the time-dependent density functional theory (TDDFT).25 Compound 1 was used to calculate two enantiomers, (1S,6S,7R,9S,10S)-1 (1c) and (1R,6R,7S,9R,10R)-1 (1d). MMFF94 conformational search and DFT re-optimization at the B3LYP/6-31+G(d) level yielded 6 lowest energy conformers for 1c (Fig. S25†). The overall calculated ECD spectra of 1c and 1d were then generated by Boltzmann weighting of the conformers (Fig. 6). The experimental ECD curve of 1 was nearly identical to the calculated ECD spectrum of 1c, suggested the 1S,6S,7R,9S,10S absolute configuration for 1, which was consistent with the result deduced from the Snatzke's method.
 | ||
| Fig. 6 Experimental ECD spectrum of 1 in MeOH and the calculated ECD spectra of 1c and 1d, after a UV correction of 10 nm. | ||
The molecular formula of pholiotin B (2) was determined to be C17H26O6 (five degrees of unsaturation) by HRESIMS, 42 mass units higher than that of 1. Analysis of the NMR spectroscopic data (Table 1) of 2 revealed nearly identical structural features to those found in 1, except that the oxymethine proton signal (H-9) was shifted downfield (δH 4.07 in 1 vs. 5.37 in 2). In addition, NMR resonances corresponding to an acetyl group (δH 2.12; δC 21.2 and 170.9) were observed, indicating that the C-9 oxygen of 2 was acylated, which was confirmed by an HMBC correlation from H-9 to the carboxylic carbon (δC 170.9) of the acetyl group. Therefore, 2 was determined as the C-9 monoacetate of 1 and its relative configuration was deduced by analogy to 1. The CD spectra of 1 and 2 (Fig. S20 and S21; ESI†) both showed positive Cotton effects at 220 nm, and negative Cotton effects at 249 nm, indicating that the absolute configuration of 2 was the same as that of 1.
Pholiotin C (3) was assigned the same molecular formula C17H26O6 as 2 by HRESIMS. Interpretation of the NMR data (Table 1) of 3 showed similar resonances to those of 2, indicating that 3 is also a monoacetate of 1 but with a different position for acetylation. Specifically, the oxymethene proton signals of H2-15 in 3 (δH 4.16, 4.22) were significantly downfield compared to 1 (δH 3.63), suggesting that OH-15 was acetylated, which was supported by an HMBC correlation from H2-15 to the carboxylic carbon of the acetyl unit (δC 171.0). On the basis of these data, 3 was determined as the C-15 monoacetate of 1. The relative and absolute configurations of 3 were deduced as shown by analogy to 1, which was further confirmed by comparison of its CD data with those of 1 (Fig. S20 and S22; ESI†).
Pholiotin D (4) gave a pseudomolecular ion [M + Na]+ peak, consistent with a molecular formula of C19H28O7 (6 degrees of unsaturation). Analysis of the 1H and 13C NMR data of 4 (Table 2) revealed that 4 is a diacetate of 1. The esterification shifts (Δ = 1.19 ppm for H-9, Δ = 0.62/0.57 ppm for H2-15) were observed, which allowed the acetyl groups to locate at C-9 and C-15, respectively. On the basis of these data, 4 was determined as the C-9 and C-15 diacetate of 1. This observation was supported by relevant HMBC correlations from H-9 to C-18 (δC 171.4) and from H2-15 to C-16 (δC 171.1). The relative and absolute configurations of 4 were deduced as shown by analogy to 1, which was further confirmed by comparison of its CD data with those of 1 (Fig. S20 and S23; ESI†).
| Pos. | 4 | 5 | ||
|---|---|---|---|---|
| δ C a | δ H b (J in Hz) | δ C a | δ H b (J in Hz) | |
| a Recorded at 125 MHz. b Recorded at 500 MHz. | ||||
| 1 | 41.6, CH | 1.83, m | 46.4, CH | 1.97, dt (12, 3.2) |
| 2a | 21.3, CH2 | 2.24, m | 34.1, CH2 | 2.28, m |
| 2b | 1.26, m | 1.49, m | ||
| 3a | 25.4, CH2 | 2.49, m | 25.5, CH2 | 2.53, dd (18, 4.6) |
| 3b | 2.18, m | 2.01, m | ||
| 4 | 131.1, qC | 131.6, qC | ||
| 5 | 139.2, CH | 7.11, s | 140.0, qC | 7.16, s |
| 6 | 39.4, CH | 2.04, m | 40.3, CH | 2.42, dt (10, 1.2) |
| 7 | 39.3, CH | 1.59, m | 38.4, CH | 1.80, m |
| 8a | 25.7, CH2 | 1.86, m | 26.7, CH2 | 1.90, dt (12, 3.3) |
| 8b | 1.46, m | 1.36, dt (3.0, 13) | ||
| 9 | 72.1, CH | 5.26, br s | 72.9, CH | 4.40, br s |
| 10 | 73.6, qC | 155.1, qC | ||
| 11 | 169.9, qC | 168.5, qC | ||
| 12 | 25.6, CH | 2.13, m | 25.5, CH | 2.28, m |
| 13 | 20.6, CH3 | 0.95, d (6.9) | 21.5, CH3 | 0.98, d (6.9) |
| 14 | 14.4, CH3 | 0.89, d (6.9) | 15.3, CH3 | 0.82, d (6.9) |
| 15a | 64.7, CH2 | 4.25, d (12) | 106.0, CH2 | 4.88, s |
| 15b | 4.21, d (12) | 4.66, s | ||
| 16 | 171.1, qC | |||
| 17 | 21.1, CH3 | 2.15, s | ||
| 18 | 171.4, qC | |||
| 19 | 21.4, CH3 | 2.12, s | ||
The elemental composition of pholiotin E (5) was established as C15H22O3 (five degrees of unsaturation) by HRESIMS. Analysis of the 1H and 13C NMR data (Table 2) of 5 revealed almost identical with those found of 1, except that the sp3 quaternary carbon (δC 74.9) and the oxymethylene (δH 3.63; δC 62.8) were replaced by the exocyclic olefin C-10/C-15 (δH 4.66 and 4.88; δC 106.0 and 155.1, respectively), which was confirmed by HMBC cross-peaks from H-6 to C-10, H-9 to C-15, and from H2-15 to C-1, C-9, and C-10. Therefore, the structure of 5 was determined and the relative configuration was confirmed by X-ray crystallography (Fig. 3).
The absolute configuration of 5 was deduced by comparison of the experimental and the simulated circular dichroism (CD) spectra (Fig. 7) generated by the time-dependent density functional theory (TDDFT).25 Compound 5 was used to calculate two enantiomers, (1R,6R,7S,9R)-5 (5a) and (1S,6S,7R,9S)-5 (5b). MMFF94 conformational search and DFT re-optimization at the B3LYP/6-31+G(d) level yielded 4 lowest energy conformers for 5a (Fig. S26†). The overall calculated ECD spectra of 5a and 5b were then generated by Boltzmann weighting of the conformers (Fig. 7). The experimental ECD curve of 5 was nearly identical to the calculated ECD spectrum of 5b, suggested the 1S,6S,7R,9S absolute configuration for 5.
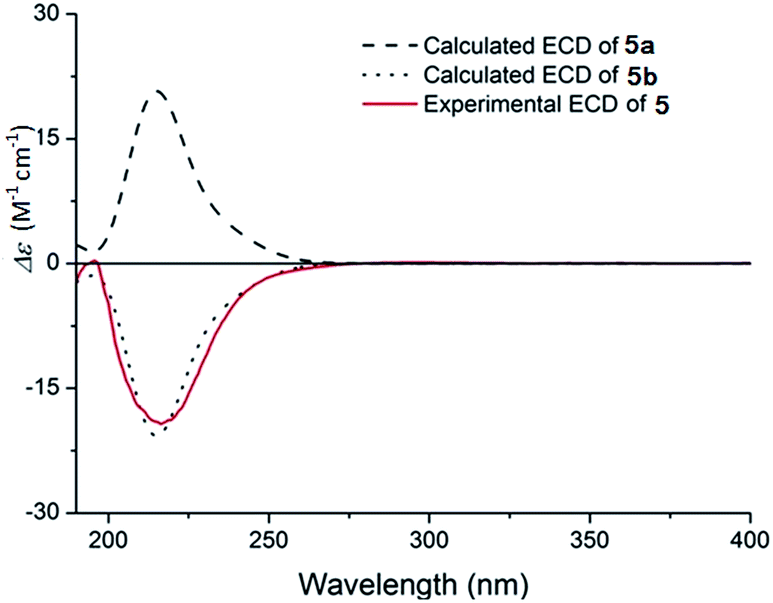 | ||
| Fig. 7 Experimental ECD spectrum of 5 in MeOH and the calculated ECD spectra of 5a and 5b, after a UV correction of 10 nm. | ||
The known compounds 6–8 isolated from the crude extract were identified as 11-hydroxy-1(10)-valencen-2-one (6),17 8,11-dihydroxy-1(10)-eremophilen-2-one (7)18 and durgamone (8),19,20 respectively, by comparison of their NMR and MS data with those reported.
Compounds 1–8 were tested for antifungal activity of Aspergillus flavus (CGMCC 3.0951), Fusarium nivale (CGMCC 3.4600) and Piricularia oryzae (CGMCC 3.3283). Compound 4 showed antifungal effects against Aspergillus flavus (CGMCC 3.0951), with an IC50 value 25.1 µM, while the positive control amphotericin B showed an IC50 value of 3.3 µM. Other compounds proved to be inactive in antifungal assays even at 200 µg mL−1.
Conclusions
In conclusion, five new cadinane sesquiterpenoids, pholiotins A–E (1–5) have been isolated from the crude extract of Pholiota sp. Their structures were elucidated based on NMR spectroscopic data and electronic circular dichroism (ECD) calculations. The activity experiments exhibited that compound 4 showed antifungal activities against Aspergillus flavus (CGMCC 3.0951). Pholiotins A–E (1–5) are new cadinane sequiterpenoids. Pholiotin A (1) is structurally related to the known fungal metabolite dysodensiol D,26 but differs by having two more hydroxy groups at C-9 and C-15, respectively. Compound 2 is an acetylation product of 1 at C-9, whereas 3 is an acetylation product of 1 at C-15. Pholiotin D (4) is the C-9 and C-15 diacetylation product of 1. Pholiotin E is a new analogue of 4,10(14)-cadinadien-15-oic acid, differing by having a hydroxy group attached to C-9.27 This was the first report that cadinane sesquiterpenoids were isolated and identified from the genus Pholiota.Experimental
General experimental procedures
Optical rotations were measured on a Perkin-Elmer 241 polarimeter, and UV data were recorded on a Shimadzu Biospec-1601 spectrophotometer. CD spectra were recorded on a JASCO J-815 spectropolarimeter using MeOH as solvent. IR data were recorded using a Nicolet Magna-IR 750 spectrophotometer. 1H and 13C NMR data were acquired with Varian Mercury-500 spectrometer using solvent signals (acetone-d6: δH 2.05/δC 29.8, 206.1; CDCl3; δH 7.26/δC 77.7) as references. The HMQC and HMBC experiments were optimized for 145.0 and 8.0 Hz, respectively. ESIMS data were recorded on a Bruker Esquire 3000plus spectrometer, and HRESIMS data were obtained using Bruker APEX III 7.0 T and APEX II FT-ICR spectrometers, respectively.Fungal material
The culture of Pholiota sp. was isolated from a soil sample on the surface of the fruiting body of C. sinensis (Berk.) Sacc. collected in Kangding, Sichuan, People's Republic of China. The isolate was identified by one of the authors (X. L.) based on morphology and sequence (Genbank Accession No. JQ411813) analysis of the ITS region of the rDNA and assigned the accession number SCK05-7-ZP19 in X. L's culture collection at the Institute of Microbiology, Chinese Academy of Sciences, Beijing. The fungal strain was cultured on slants of potato dextrose agar (PDA) at 25 °C for 10 days. Agar plugs were cut into small pieces (about 0.5 × 0.5 × 0.5 cm3) under aseptic conditions, 15 pieces were used to inoculate three Erlenmeyer flasks (250 mL), each containing 50 mL of media (0.4% glucose, 1% malt extract, and 0.4% yeast extract); the final pH of the media was adjusted to 6.5 and sterilized by autoclave. Three flasks of the inoculated media were incubated at 25 °C on a rotary shaker at 170 rpm for five days to prepare the seed culture. Fermentation was carried out in eight Fernbach flasks (500 mL), each containing 80 g of rice. Spore inoculum was prepared by suspension in sterile, distilled H2O to give a final spore/cell suspension of 1 × 106 mL. Distilled H2O (120 mL) was added to each flask, and the contents were soaked overnight before autoclaving at 15 psi for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the spore inoculum and incubated at 25 °C for 40 days.Extraction and isolation
The fermented material was extracted repeatedly with EtOAc (4 × 1.0 L), and the organic solvent was evaporated to dryness under vacuum to afford the crude extract (3.1 g), which was fractionated by silica gel VLC using petroleum ether–EtOAc gradient elution. The fraction (62 mg) eluted with 50% EtOAc was separated by Sephadex LH-20 column chromatography (CC) eluting with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2–MeOH. The resulting subfractions were combined and further purified by RP HPLC (Agilent Zorbax SB-C18 column; 5 µm; 9.4 × 250 mm; 42% MeOH in H2O for 5 min, followed by 45–100% for 30 min; 2 mL min−1) to afford 1 (5.2 mg, tR 17.32 min) and 4 (2.0 mg, tR 26.21 min). The fraction (32 mg) eluted with 60% EtOAc was also separated by RP HPLC (60% MeOH in H2O for 5 min, followed by 80–90% for 25 min) to afford 5 (2.1 mg, tR 15.23 min). The fraction (98 mg) eluted with 65% EtOAc was also separated by RP HPLC (50% MeOH in H2O for 5 min, followed by 80–90% for 35 min) to afford 6 (3.2 mg, tR 17.28 min), 7 (2.8 mg, tR 28.20 min) and 8 (1.5 mg, tR 20.30 min). The fraction (90 mg) eluted with 70% EtOAc was purified by RP HPLC (42% MeOH in H2O for 5 min, followed by 45–100% for 30 min) to afford 2 (4.5 mg, tR 18.94 min) and 3 (3.6 mg, tR 21.12 min).
:1 CH2Cl2–MeOH. The resulting subfractions were combined and further purified by RP HPLC (Agilent Zorbax SB-C18 column; 5 µm; 9.4 × 250 mm; 42% MeOH in H2O for 5 min, followed by 45–100% for 30 min; 2 mL min−1) to afford 1 (5.2 mg, tR 17.32 min) and 4 (2.0 mg, tR 26.21 min). The fraction (32 mg) eluted with 60% EtOAc was also separated by RP HPLC (60% MeOH in H2O for 5 min, followed by 80–90% for 25 min) to afford 5 (2.1 mg, tR 15.23 min). The fraction (98 mg) eluted with 65% EtOAc was also separated by RP HPLC (50% MeOH in H2O for 5 min, followed by 80–90% for 35 min) to afford 6 (3.2 mg, tR 17.28 min), 7 (2.8 mg, tR 28.20 min) and 8 (1.5 mg, tR 20.30 min). The fraction (90 mg) eluted with 70% EtOAc was purified by RP HPLC (42% MeOH in H2O for 5 min, followed by 45–100% for 30 min) to afford 2 (4.5 mg, tR 18.94 min) and 3 (3.6 mg, tR 21.12 min).
Pholiotin A (1)
White needles, (acetone–H2O), mp 230–235 °C; [α]25D +15.7 (c 0.6, MeOH); UV (MeOH) λmax (logε) 212 (3.39), 227 (3.40) nm; CD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 220 (+2.7), 249 (−1.9); IR (neat) νmax 3334, 2953, 1710, 1640, 1408, 1278, 1069, 1040 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 500 MHz) H-1 → C-2, 3, 6, 7, 10, 15; H2-2 → C-1, 3, 4, 6; H-3b → C-11; H-5 → C-1, 3, 4, 7, 11; H-6 → C-2, 4, 8, 12; H-7 → C-8; H2-8 → C-6, 10, 12; H-12 → C-7, 8, 13, 14; H3-13 → C-7, 12, 14; H3-14 → C-7, 12, 13; H2-15 → C-1, 9, 10; HREIMS m/z 307.1513 (calcd for C15H24O5Na 307.1516).
X-ray crystallographic analysis of pholiotin A (1)28
Upon crystallization from acetone–H2O (5:1) using the vapor diffusion method, colorless crystals were obtained for 1, a crystal (0.23 × 0.14 × 0.12 mm) was separated from the sample and mounted on a glass fiber, and data were collected using a Rigaku RAPID IP diffractometer with graphite-monochromated Mo Kα radiation, λ = 0.71073 Å at 173(2) K. Crystal data: C15H14O5, M = 284.34, space group orthorhombic, P2(1)2(1)2(1); unit cell dimensions a = 5.5709 (11) Å, b = 15.994(3) Å, c = 16.547(3) Å, V = 1474.4(5) Å3, Z = 4, Dcalcd = 1.281 mg m−3, µ = 0.095 mm−1, F(000) = 616. The structure was solved by direct methods using SHELXL-977 (ref. 29) and refined by using full-matrix least-squares difference Fourier techniques. All non-hydrogen atoms were refined with anisotropic displacement parameters and all hydrogen atoms were placed in idealized positions and refined as riding atoms with the relative isotropic parameters. Absorption corrections were performed using the Siemens Area Detector Absorption Program (SADABS).30 The 13237 measurements yielded 1947 independent reflections after equivalent data were averaged, and Lorentz and polarization corrections were applied. The final refinement gave R1 = 0.0450 and wR2 = 0.1089 [I > 2σ(I)].
Preparation of (R)-(1a)
A sample of 1 (1.0 mg, 0.004 mmol), (S)-MTPA Cl (2.0 µL, 0.011 mmol), and pyridine-d5 (0.5 mL) were allowed to react in an NMR tube at ambient temperature for 24 h, with the 1H NMR data of the R-MTPA ester derivative (1a) were obtained directly on the reaction mixture: 1H NMR (pyridine-d5, 600 MHz) δ 6.99 (1H, s, H-5), 4.25 (1H, d, J = 10 Hz, H-15a), 4.21 (1H, d, J = 10 Hz, H-15b), 3.82 (1H, s, H-9), 2.37 (1H, dd, J = 15, 5.7 Hz, H-3a), 2.13 (1H, td, J = 5.5, 1.0 Hz, H-2a), 2.11 (1H, m, H-12), 2.07 (1H, m, H-3b), 2.00 (1H, m, H-6), 1.73 (1H, t, J = 11 Hz, H-8a), 1.68 (1H, dt, J = 12, 3.3 Hz, H-1), 1.52 (1H, m, H-7), 1.33 (1H, dt, J = 11, 1.5 Hz, H-8b), 1.13 (1H, m, H-2b), 0.90 (3H, d, J = 6 Hz, H3-13), 0.76 (3H, d, J = 5.5 Hz, H3-14).Absolute configuration of the 9,10-diol moiety in 1a23,24
HPLC grade DMSO was dried with 4 Å molecular sieves. According to a published procedure, a mixture of 1:1.3 diol/Mo2(OAc)4 for 1a was subjected to CD measurements at a concentration of 1.0 mg mL−1. The first CD spectrum was recorded immediately after mixing, and its time evolution was monitored until stationary (about 10 min after mixing). The inherent CD was subtracted. The observed signs of the diagnostic bands at around 310 and 400 nm in the induced CD spectrum were correlated to the absolute configuration of the 9,10-diol moiety.
Pholiotin B (2)
White powder; [α]25D +20.5 (c 0.4, MeOH); UV (MeOH) λmax (logε) 216 (3.39), 220 (3.38) nm; CD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 222 (+2.2), 249 (−0.8); IR (neat) νmax 3413, 2957, 1692, 1641, 1371, 1243, 1039 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 500 MHz) H-1 → C-2, 3, 6, 9; H2-2 → C-1, 3, 6; H2-3 → C-1, 2, 4, 5; H-5 → C-1, 3, 7, 11; H-6 → C-1, 2, 4, 8, 12; H-7 → C-6, 8; H2-8 → C-6, 7, 12; H-9 → C-1, 7, 10, 18; H-12 → C-7, 13, 14; H3-13 → C-7, 12, 14; H3-14 → C-7, 12, 13; H2-15 → C-1, 9, 10; H3-19 → C-18; HRESIMS m/z 349.1619 (calcd for C17H26O6Na, 349.1622).
Pholiotin C (3)
White powder; [α]25D +26.9 (c 0.4, MeOH); UV (MeOH) λmax (logε) 216 (3.55), 220 (3.62) nm; CD (c 1.3 × 10−3 M, MeOH) λmax (Δε) 219 (+2.7), 250 (−0.8); IR (neat) νmax 3422, 2956, 1690, 1641, 1371, 1241, 1040 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 500 MHz) H-1 → C-3, 15; H-2a → C-1, 6; H-2b →C-1, 3; H2-3 → C-1, 2, 4, 5; H-5 → C-3, 4, 6, 7, 11; H-7 → C-8; H2-8 → C-6, 10; H-9 → C-7; H-12 → C-7, 13, 14; H3-13 → C-7, 12, 14; H3-14 → C-7, 12, 13; H2-15 → C-1, 9, 10, 16; H3-17 → C-16; HREIMS m/z 349.1623 (calcd for C17H26O6Na 349.1622).
Pholiotin D (4)
White powder; [α]25D +15.5 (c 0.2, MeOH); UV (MeOH) λmax (logε) 212 (3.14), 220 (3.13) nm; CD (c 0.8 × 10−3 M, MeOH) λmax (Δε) 218 (+0.5), 259 (−0.2); IR (neat) νmax 3440, 2931, 1719, 1372, 1239, 1037 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 500 MHz) H-1 → C-3, 15; H2-2 → C-6; H2-3 → C-1; H-5 → C-1, 11; H-6 → C-2; H-7 → C-8; H2-8 → C-6, 10, 12; H-9 → C-1, 7, 10, 18; H-12 → C-7, 13, 14; H3-13 → C-7, 12, 14; H3-14 → C-7, 12, 13; H2-15 → C- 9, 16; H3-17 → C-16; H3-19 → C-18; HRESIMS m/z 391.1728 (calcd for C19H28O7Na, 391.1727).
Pholiotin E (5)
White needle; mp 130–135 °C; [α]25D −16.9 (c 0.2, MeOH); UV (MeOH) λmax (logε) 214 (3.94) nm; CD (c 1.0 × 10−3 M, MeOH) λmax (Δε) 217 (−20); IR (neat) νmax 3322, 2950, 2921, 2624, 1689, 1643, 1390, 1259, 1057 cm−1; 1H and 13C NMR data see Table 1; HMBC data (acetone-d6, 500 MHz) H2-2 → C-4; H2-3 → C-1, 5; H-5 → C-1, 3, 11; H-6 → C-2, 10; H-7 → C-1; H-9 → C-1, 7, 15; H-12 → C-7, 13, 14; H3-13 → C-7, 12, 14; H3-14 → C-7, 12, 13; H2-15 → C-1, 9, 10; HRESIMS m/z 249.1498 (calcd for C15H21O3, 249.1496).
X-ray crystallographic analysis of pholiotin E (5)31
Upon crystallization from acetone–H2O (5:1) using the vapor diffusion method, colorless crystals were obtained for 5, a crystal (0.20 × 0.15 × 0.10 mm) was separated from the sample and mounted on a glass fiber, and data were collected using a Rigaku RAPID IP diffractometer with graphite-monochromated Mo Kα radiation, λ = 0.71073 Å at 173(2) K. Crystal data: C30H46O7, M = 518.67, space group orthorhombic, P2(1)2(1)2; unit cell dimensions a = 16.315 (3) Å, b = 17.050 (3) Å, c = 5.2386 (10) Å, V = 1457.3 (5) Å3, Z = 2, Dcalcd = 1.182 mg m−3, µ = 0.083 mm−1, F(000) = 564. The structure was solved by direct methods using SHELXL-97 (ref. 29) and refined by using full-matrix least-squares difference Fourier techniques. All non-hydrogen atoms were refined with anisotropic displacement parameters and all hydrogen atoms were placed in idealized positions and refined as riding atoms with the relative isotropic parameters. Absorption corrections were performed using the Siemens Area Detector Absorption Program (SADABS).30 The 12011 measurements yielded 1929 independent reflections after equivalent data were averaged, and Lorentz and polarization corrections were applied. The final refinement gave R1 = 0.0430 and wR2 = 0.1026 [I > 2σ(I)].
11-Hydroxy-1(10)-valencen-2-one (6)
1H, 13C NMR, and the MS data were consistent with literature values.178,11-Dihydroxy-1(10)-eremophilen-2-one (7)
1H, 13C NMR, and the MS data were consistent with literature values.18Durgamone (8)
1H, 13C NMR, and the MS data were consistent with literature values.19,20Computational details
Systematic conformational analyses for 1c and 1d, 5a and 5b were performed via the Molecular Operating Environment (MOE) ver. 2009.10. (Chemical Computing Group, Canada) software package using the MMFF94 molecular mechanics force field calculation. The MMFF94 conformational analyses were further optimized using DFT at the B3LYP/6-31+G(d) or cam-B3LYP/6-31G(d) basis set level. The stationary points have been checked as the true minima of the potential energy surface by verifying they do not exhibit vibrational imaginary frequencies. The 50 lowest electronic transitions were calculated at the Cam-B3LYP/6-31+G(d) level, and the rotational strengths of each electronic excitation were given using both dipole length and dipole velocity representations. ECD spectra were stimulated using a Gaussian function with a half-bandwidth of 0.3 eV. Equilibrium populations of conformers at 298.15 K were calculated from their relative free energies (ΔG) using Boltzmann statistics. The overall ECD spectra were then generated according to Boltzmann weighting of each conformer. The systematic errors in the prediction of the wavelength and excited-state energies are compensated for by employing UV correction.25Antifungal assays
Antifungal assays were conducted in triplicate following the National Center for Clinical Laboratory Standards (NCCLS) recommendations.32 The fungi, Aspergillus flavus (CGMCC 3.0951), Piricularia oryzae (CGMCC 3.3283), and Fusarium nivale (CGMCC 3.4600) were obtained from China General Microbial Culture Collection (CGMCC) and were grown on PDA. Targeted fungi (3–4 colonies) were prepared from broth culture (A. flavus: 28 °C for 36 h; the plant pathogens: 28 °C for 48 h) and the final suspensions contained 104 hyphae per mL (in PDB medium). Test samples (4 mg mL−1 as stock solution in DMSO and serial dilutions) were transferred to 96-well clear plate in triplicate, and the suspensions of the test organisms were added to each well, achieving a final volume of 200 µL. Alamar blue (10 µL of 10% solution) was added to each well as an indicator and amphotericin B and carbendazim were used as the positive controls. After incubation (A. flavus: 28 °C for 36 h; the plant pathogens: 28 °C for 48 h), the fluorescence intensity was measured at Ex/Em = 544/590 nm. The inhibition was calculated and plotted versus test concentrations to afford the IC50.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21372004), the Program of the Excellent Young Scientists of Chinese Academy of Sciences, the Youth Innovation Promotion Association of Chinese Academy of Sciences (2011083) and Natural Science Fund of Colleges and Universities in Jiangsu Province (16KJA180005, 16KJB180006).Notes and references
- J. B. Gloer, Acc. Chem. Res., 2002, 28, 343–350 CrossRef.
- X. L. Feng, Y. Yu, H. Gao, Z. Q. Mu, X. R. Cheng, W. X. Zhou and X. S. Yao, RSC Adv., 2014, 46, 42071–42077 RSC.
- Q. Shou, J. E. Smith, H. Mon, Z. Brkljaca, A. S. Smith, D. M. Smith, H. J. Griesser and H. Wohlmuth, RSC Adv., 2014, 4, 13514–13517 RSC.
- P. Zhang, L. H. Meng, A. Mandi, X. M. Li, T. Kurtan and B. G. Wang, RSC Adv., 2015, 46, 39870–39877 RSC.
- M. M. Dreyfuss and I. H. Chapela, Biotechnology, 1994, 26, 49–80 CAS.
- A. Jegorov, P. Šimek, A. Heydová, L. Cvak and J. Minář, Applications of Fungal Ecology in the Search for New Bioactive Natural Products, Springer, Berlin, Heidelberg, 2007, pp. 249–268 Search PubMed.
- H. Guo, H. Hu, S. Liu, X. Liu, Y. Zhou and Y. Che, J. Nat. Prod., 2007, 70, 1519–1521 CrossRef CAS PubMed.
- Y. Zhang, S. Liu, Y. Che and X. Liu, J. Nat. Prod., 2007, 70, 1522–1525 CrossRef CAS PubMed.
- Y. Zhang, S. Liu, H. Liu, X. Liu and Y. Che, J. Nat. Prod., 2009, 72, 1364–1367 CrossRef CAS PubMed.
- C. Ma, Y. Li, S. Niu, H. Zhang, X. Liu and Y. Che, J. Nat. Prod., 2011, 74, 32–37 CrossRef CAS PubMed.
- J. Lin, S. Liu, B. Sun, S. Niu, E. Li, X. Liu and Y. Che, J. Nat. Prod., 2010, 73.0, 905–910 CrossRef PubMed.
- J. Lin, X. Chen, X. Cai, X. Yu, X. Liu, Y. Cao and Y. Che, J. Nat. Prod., 2011, 74, 1798–1804 CrossRef CAS PubMed.
- T. Nakazawa and T. Tochigi, Chest, 1989, 95, 1149–1151 CrossRef CAS PubMed.
- K. Konishi, T. Mouri, Y. Kojima, E. Chida, K. Sugawara, K. Abe, T. Bando, M. Ishii and M. Tamura, Nihon Kyobu Shikkan Gakkai Zasshi, 1994, 32, 655–661 CAS.
- M. Ishii, A. Kikuchi, K. Kudoh, K. Konishi, T. Mohri, M. Tamura and N. Tomichi, Intern. Med., 1994, 33, 683–685 CrossRef CAS PubMed.
- M. Inage, H. Takahashi, H. Nakamura, I. Masakane and H. Tomoike, Intern. Med., 1996, 35, 301–304 CrossRef CAS PubMed.
- G. Savona, F. Piozzi, M. C. D. L. Torre, O. Servettaz and B. Rodríguez, Phytochemistry, 1987, 26, 571–572 CrossRef CAS.
- M. Park, W. Fenical and M. E. Hay, Phytochemistry, 1992, 31, 4115–4118 CrossRef CAS.
- D. Soorukram, T. Qu and A. G. M. Barrett, Org. Lett., 2008, 10, 3833–3835 CrossRef CAS PubMed.
- A. Rudi, T. Yosief, M. Schleyer and Y. Kashman, Tetrahedron, 1999, 55, 5555–5566 CrossRef CAS.
- J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092–4096 CrossRef CAS.
- M. Górecki, E. Jabłońska, A. Kruszewska, A. Suszczyńska, Z. Urbańczyk-Lipkowska, M. Gerards, J. W. Morzycki, W. J. Szczepek and J. Frelek, J. Org. Chem., 2007, 72, 2906–2916 CrossRef PubMed.
- B. L. Di, G. Pescitelli, C. Pratelli, D. Pini and P. Salvadori, J. Org. Chem., 2001, 66, 4819–4825 CrossRef.
- N. Berova, L. D. Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- B. J. Xie, S. P. Yang and J. M. Yue, Phytochemistry, 2008, 69, 2993–2997 CrossRef CAS PubMed.
- F. Bohlmann, C. Zdero, J. Pickard, H. Robinson and R. M. King, Phytochemistry, 1981, 20, 1323–1333 CrossRef CAS.
- Crystallographic data for 1 have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 861573†).
- G. M. Sheldrick, SHELXL-97, Program for X-ray Crystal Structure Solution and Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Göttingen, Germany, 1999 Search PubMed.
- Crystallographic data for 5 have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 861574†).
- NCCLS, Reference method for broth dilution antifungal susceptibility testing of filamentous fungi; approved standard, NCCLS document M38-A, NCCLS, Wayne, PA, 2002 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR spectra of compounds 1–5. CCDC 861573 and 861574. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra22448b |
| This journal is © The Royal Society of Chemistry 2016 |