
N-Alkylation of amines with phenols over highly active heterogeneous palladium hydride catalysts†
Abstract
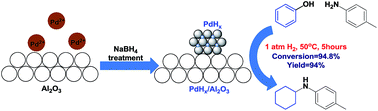
Phenols are directly converted to secondary amines in considerable yield via hydrogenation and amination tandem reaction over Al2O3 supported palladium hydride (PdHx) bi-functional catalyst. Note that this system proceeds efficiently with mild conditions under H2 atmosphere, which was difficult to achieve in previous reports. The catalyst and the mechanism of reaction are both studied. Furthermore, various secondary amines can be formed in good yields under this conversion system.
 Please wait while we load your content...
Please wait while we load your content...

